|
|
CAROL
Mutagenesis Primers Set Up
Purpose: To set up primers at the correct concentration to perform the QuikChange XL Site-Directed Mutagenesis.
- Primers have to be set up to have a concentration of 125 ng/µL. The primers were ordered from the synthesis lab at the University of Calgary
The following are the concentrations:
| Primer | 260/280 | 260/230 | Concentration [ng/μL] | Reading/2.5 [ng/μL]
|
| ''luxCDABE-M-F | 1.52 | 2.33 | 409.5 | 163.8
|
| ''luxCDABE-M-R | 1.58 | 2.86 | 479.8 | 191.2
|
- Checked if we have NZY+ broth prepared (found in freezer).
|
|
|
EMILY
Construction Digest to Move From pCR2.1-TOPO to psB1AC3 Vector
- Objective: The LuxOD47E gene in now in linear form with the Biopbrick restriction sites attatched. We now want to get it into the psB1AC3 vector,
- Protcol: To do this, we will digest both the gene and the vector in two ways: one way with EcoRI and PstI and one way with XbaI and PstI
- Insert tube 1- DNA to approximately [600ng/uL]= 3.1uL
ddH2O up to 35 uL- 31.9 uL
4 uL REact 2 Buffer
1 uL XbaI restriction enzyme
1 uL PstI restriction enzyme
- Insert tube 2- same as for tube one, but with EcoRI restriction enzyme in place of XbaI restriction enzyme.
- Vector tube 1- psB1AC3 to [200ng/uL], REact 2 Buffer, XbaI restriction enzyme and PstI restriction enzyme
- Vector tube 2- same as for tube one, but with EcoRI restriction enzyme in place of XbaI restriction enzyme.
- Placed in 37 C waterbath for 2,5 hours followed by heat deactivation for 10 minutes in a 65 C heating block.
|
|
|
FAHD
Marketing for June 16th 2009
NUTV (New University Television) is a non-profit organization that seeks to provide University of Calgary students with the means to participate in television production. NUTV programs are aired on campus and in Calgary. This year, iGEM Calgary has had the opportunity to appear on NUTV as a part of our 2009 media campaign. We were part of its monthly show called Full Frontal, a magazine-styled segment showcasing news, art, and human interest stories. The iGEM Calgary segment was aired on July 4th. 2009. This story was covered by the NUTV news Reporter Julie Phillips and edited by the NUTV programme director Justin Hardjowirogo. The news coverage explored the different areas of our 2009 iGEM Team: namely Wetlab, Mathematical Modeling, Second Life (TM) and Ethics.
We also introduced the iGEM competition, synthetic biology, and our quorum sensing project. The video included interviews from Thane Alexander Kubik (iGEM Calgary Team Co-Leader), Carol Chan (Engineering Student) and Patrick King (Second Life Team Leader).
|
|
|
JAMIE
Construction verification via colony PCR
Colonies grew on the plates! Verification with colony PCR using pTaq (Invitrogen, CA). Forward and reverse gene specific primers (Tm: 60oC). Standard manufacturer's directions followed.
Restreaks of colonies.
|
|
|
JEREMY
Colony PCR of LuxPQ in psB1AK3
Seven colonies were screened with pTaq colony PCR of LuxPQ in psB1AK3 using luxPQ F/R primers and the following conditions: 94ºC for 6 minutes; 30 X (94ºC for 30 seconds; 55ºC for 45 seconds; 72ºC for 4 minutes); 72ºC for 10 minutes; held at 4ºC.
|
|
|
KATIE
A Display for Quorum Sensing
I have returned to the avatar I made to sense a specific object and decided to use it to create a primitive quorum sensing system. To do so, it can no longer use the collision event in order to detect the presence of another object, but will have to use a sensor instead. I have decided to abandon the script to attach the object to a user’s original avatar and work on getting the sensor event to work for me. This will involve:
- Creating a variable to keep track of how many of the same objects are around each other. This may be determined by detecting the names of the objects around them
- Determining the range of the sensor so that the bacteria do not sense each other so easily that the point of what they are doing is lost
- Have the bacteria glow in response to a threshold level of bacteria. I will most likely have the bacteria as a display so I would like to keep them within a certain area, most likely in a small section of the synthetic kingdom
- Organize an explanatory note card for the quorum sensing bacteria
I have also begun planning out a restriction digest activity, which I will continue to work on tomorrow.
|
|
|
KEVIN
Modelling
Researched on the reaction rates of various reactions, such as the binding affinity of AI-2 to LuxP.
|
|
|
MANDY
Autoclaving in Second Life
Built an autoclave in Second Life and wrote a notecard to demonstrate the preparation of LB plates in lab. We didn't want to make this an activity as we felt that there are more crucial lab techniques that we can teach. Took some more photos of the lab for building/sculpting and texturing purposes.
|
|
|
PATRICK
TetR is A Dimer
Began working on 'dimerizing' subunits of a protein. I had wanted both halves of any given protein to be identical in game (as they are in world), and not artificially choose one to be the 'dominant' or 'main' part, that manages all of the interactions for its complex. This turned out to be very complicated, and ultimately not worth the effort. Realistically portraying dimers in game is great, but it adds an extra step between elements of the simulator that we actually care about: production of regulators, and their action on different dna elements. This is especially the case for something like CI Lambda, which can form dimers, tetramers, and even octamers in different circumstances! Portraying the realism at *that* level would overload the user with pointless tasks, for no real gain.
Other pieces of work done today, first began experimenting with putting CSV (comma separated values) information in the names of objects (such as DNA, to record what it is, what it should allow to bind with it, what its neighbours name is. Also noticed that once bound there would be no good way to unbind objects from each other. The only easy interactions the user can have with objects are clicking them and sending messages to them. I'd already used clicking for movement, and the last thing I wanted to do was force people to memorize some messaging scheme. This was the need that lead to the 'unbind' screen.
Ultimately, I produced a working set of dimerizing TetR molecules, that worked to repress a promoter and prevent RNAP from binding. But gene expression, and the ability to unbind that TetR molecule, would be a long time coming.
|
|
|
PRIMA
Shadowed Jeremy in Colony PCR & Restriction Digest
Purpose of Colony PCR: to see if the gene size that we constructed has been uptaken by the bacteria.
Method: please refer to protocol page on the wiki.
We used Lux PQ-F/R primers.
Note to self: pTaq DNA polymerase is always added last.
Purpose of RD: To verify the size of the Lux PQ construct
With digested with EcoRI +PstI in tubes 1-6 and with XbaI + Pst in tube 6 and tube 8 was the negative control.
Before running on gel:
-add 5 microL PCR product
-add 2 microL orange dye
- add 15 microL ddH2O
so that the total volume is 20 microL.
Then I helped to make the 0.8% agarose gel. We ran the digest at 90V.
|
|
|
VICKI
Colony PCR of LuxOD47A BBk
Purpose: To isolate the colonies in which LuxOD47A on psB1AC3 was successfully transformed
Protocol:
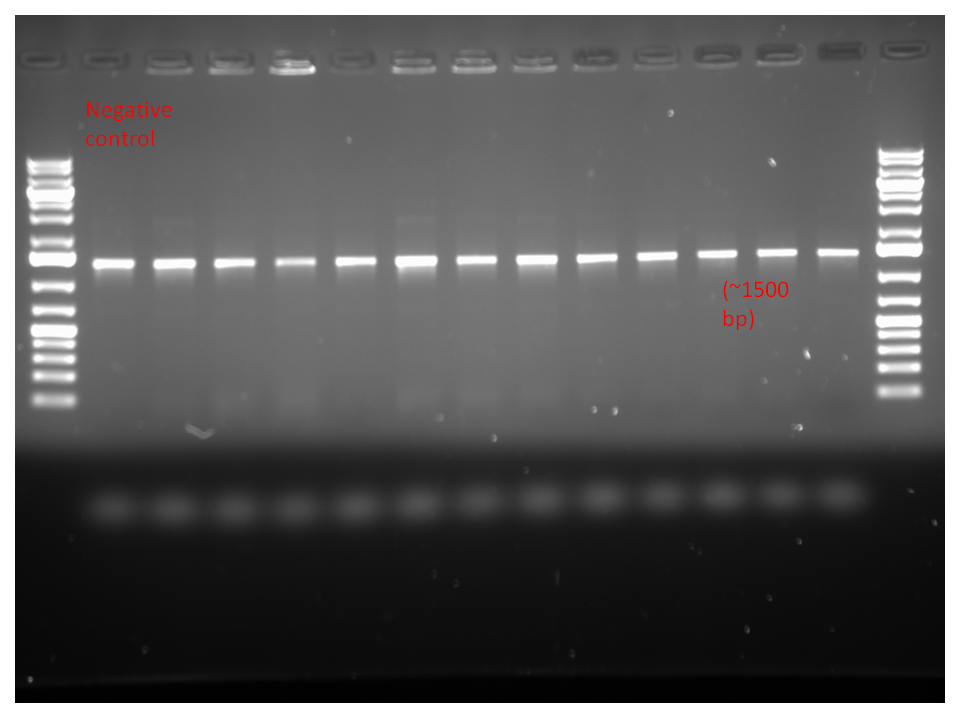
We conducted a colony PCR of the colonies that grew on the overnight plates as a preliminary verification that our LuxOD47A BBk sequences on pSB1AC3 plasmids were properly transformed into the cells. We prepared tubes for the six largest colonies from each sample (initially formed with EcoRI and XbaI), for a total of twelve colony PCR tubes. As this process is only for verification (as opposed to LuxOD47A BBk formation from the gradient PCR with gene-specific BBk primers), the high fidelity afforded by the Platinum Pfx polymerase is not necessary, so the less-expensive and sufficiently-reliable Platinum Taq polymerase - and the corresponding Taq PCR buffer, with MgCl2 instead of MgSO4, was used instead in the Master Mix. Forward and reverse LuxOD47A-specific primers (without BioBrick sites) were used in the sequence amplification. We completed the Master Mix by adding dNTPs and double-distilled water, and then evenly distributed 650 uL of it across thirteen tubes (twelve for the colony testing and one negative control), each containing 50 uL aliquots of Master Mix. To add DNA to our relevant tubes, we delicately touched a pipette tip to a colony of interest and immediately inserted the bacteria-infested tip into the respective PCR tube. This was done for every tube except the negative control, which only contained Master Mix.
We then proceeded to run the colony PCR. Unlike the gradient PCR, the annealing temperature does not vary between wells and is set at 5 degrees C below the primer annealing temperature of 60 degrees C. The purpose of each step in the process is consistent, however. The Platinum Taq polymerase was activated in a 6 minute initialisation step at 94 degrees C. 36 cycles of a denaturation step (30 seconds at 94 degrees C), an annealing step (45 seconds at 55 degrees C) and an extension step (90 seconds at 72 degrees C). This was followed by a 10 minute final extension at 72 degrees C, and held at 4 degrees C once the PCR was complete.
Results: These are shown below. The bands are an appropriate size, but the negative control lane is not distinct from the other lanes. We progressed anyway with our restriction digest verification. Restreaks were also made for each of the colonies that were tested in the colony PCR.
|
 "
"











