 "
"
 Project Report
Project Report
Contents
|
The goal of providing an universal restriction enzyme was approached
with two design strategies. The first strategy evolves around novel
protein fusion constructs combining the cleavage domain of the Type IIs
restriction enzyme FokI with hapten binding anticalins, which are then
guided to their target sites by a modified oligonucleotide. The second
approach aimed at converting the Argonaute proteins from thermophilic
organisms from an RNase to an DNase activity while accepting a DNA
guide oligonucleotide. For the Fok-based strategy, several variations
were tested. In both projects important milestones were reached. Most
importantly, we demonstrated guided cleavage by one of our Fok-based
fusion constructs and phage display of an Argonaute protein.
In the following we introduce these two projects and
then list the highlights of our work with links to detailed
descriptions of each project part.
Both strategies rely on locating the universal restriction enzyme at the cleavage site with adapter or guide oligonucleotides. This is in contrast to previous designs which either use chemical linkage of an oligonucleotide to a nuclease or genetic fusion with a DNA binding domain. Previous fusion protein approaches have the conceptual disadvantage that for each target sequence a special protein has to be designed, expressed, purified, and stored. We only need to produce one protein. The oligonucleotide planning is aided by readily available PCR primer design programs and there are virtually no limits to define the cleavage site. Modified oligonucleotides can be ordered from many suppliers at low cost with short delivery times and easily can be stored long term. In in-vitro applications hybridization of the oligonucleotide is easily achieved also to double strand by heating and cooling as it is well known from PCR procedures. In case of the Argonaute proteins from thermophiles we can assume that they survive several cycles of heat dissociation and annealing. In case of the Fok we plan to introduce thermostability by directed evolution. In addition, other groups are working with oligonucleotides forming triple helices and their general hybridization with any sequence or with peptide nucleotide acids which provide higher stability and sneak in existing double helices. These technologies are compatible with our approach.
In both of our universal restriction enzyme strategies we do not cut the double strand but rather nick the stand opposite to our guide oligonucleotide. Thus, our guide oligonucleotide only needs to be added only in catalytic quantities. The nicking feature is already present in the Argonaute proteins. In the case of the universal Fok-based enzyme we use a heterodimer design combined with cleavage inactivating mutations for one monomer.
Details of the universal restriction enzyme based on Fok-Anticalin fusions
The restriction endonuclease FokI from Flavobacterium okeanokoites is a well studied protein. It consists of two domains, a DNA recognition domain and a DNA cleavage domain. Upon recognition of one target site and dimerization it cleaves the DNA nine bases apart from the recognition site. Several groups reported experiments to uncouple the cleavage and restriction domains of FokI and created a novel site-specific endonucleases by linking the cleavage domain to zinc finger proteins (Miller et al. 2007).
For our project we combined two previous research results and generated a Fok cleavage heterodimer comprising an enzymatically active and inactive monomer. For the catalytic active Fok partner, named Fok_a, as well as for the catalytic inactive Fok partner, Fok_i, the association interface was mutated to disfavor homodimerization and promote heterodimerization. In Fok_i additional amino acid exchanges led to the inactivation.
The two heterodimeric partners were fused to anticalins binding different adapter molecules. Fok_a is genetically fused to a digoxigenin-binding anticalin (DigA) and Fok_i to a fluorescein-binding anticalin (FluA). The adapter molecules digoxigenin and fluorescein are common modifications linked to oligonucleotides thus mediating the binding to the DNA site of interest. Two modifications allow for a better spatial control of the cleavage site. On the target site Fok_i and Fok_a constructs are brought into close proximity and can form a heterodimer. The inactive Fok domain will serve as an activator of the active Fok domain, which cuts one strand of the DNA. Our structural ‘3D’ models, indicate that Fok domains can be positioned in such a way that Fok_a will cleave the target DNA, and Fok_i would be directed towards the modified oligonucleotide. Different linkers were designed and fused between cleavage domain and the anticalin binding moieties to test for the optimal distance. In addition we generated a monomeric Fok fusion construct, to enable phage display and te4st a further setting requiring only one binding domain and hapten. Furthermore, we made a Fok cleavage domain fusion with a coiled coil based DNA binding domain, because we can combine these with existing light switchable inhibitors, which prevent DNA binding. As a result we will obtaina light switchable restriction enzyme.
Milestones
To reach our goal within the short given time frame we started several subprojects in parallel. Our subprojects listed here are defined along these projects.
Designing of the constructs was aided by extensive model building and analysis of the spatial orientation of the different proteins and oligonucleotides used. All designed and constructed parts feature full BioBrick compatibility and in addition allow for the construction of fusion proteins based on the RFC 25 (Freiburg) cloning standard. Cloning of the respective parts was followed by expression purification and analysis. Despite previous literature data our active Fok construct was toxic to cells when expressed and we tested periplasmic expression with export of the nascent polypeptide chain before folding.
In our experiments we addressed the following questions:
• Structural Model building
• Design of protein fusion parts
• Cloning of anticalin Fok fusions
• Cloning of a monomeric Fok construct and of a Jun/Fos directed Fok construct
• Expression and purification of constructs
• In vitro assays
• In vivo assays
• Phage Display of an Ago protein
• Modeling of assembly and cleavage with differential equations
• An international survey of laymen on synthetic biology
In short, we successfully worked on all aspects. Experiments to validate our approach in more compelx settings are ongoing.
The labs of Kristian Müller and Katja Arndt provided all technology and support for the project.
Both strategies rely on locating the universal restriction enzyme at the cleavage site with adapter or guide oligonucleotides. This is in contrast to previous designs which either use chemical linkage of an oligonucleotide to a nuclease or genetic fusion with a DNA binding domain. Previous fusion protein approaches have the conceptual disadvantage that for each target sequence a special protein has to be designed, expressed, purified, and stored. We only need to produce one protein. The oligonucleotide planning is aided by readily available PCR primer design programs and there are virtually no limits to define the cleavage site. Modified oligonucleotides can be ordered from many suppliers at low cost with short delivery times and easily can be stored long term. In in-vitro applications hybridization of the oligonucleotide is easily achieved also to double strand by heating and cooling as it is well known from PCR procedures. In case of the Argonaute proteins from thermophiles we can assume that they survive several cycles of heat dissociation and annealing. In case of the Fok we plan to introduce thermostability by directed evolution. In addition, other groups are working with oligonucleotides forming triple helices and their general hybridization with any sequence or with peptide nucleotide acids which provide higher stability and sneak in existing double helices. These technologies are compatible with our approach.
In both of our universal restriction enzyme strategies we do not cut the double strand but rather nick the stand opposite to our guide oligonucleotide. Thus, our guide oligonucleotide only needs to be added only in catalytic quantities. The nicking feature is already present in the Argonaute proteins. In the case of the universal Fok-based enzyme we use a heterodimer design combined with cleavage inactivating mutations for one monomer.
Details of the universal restriction enzyme based on Fok-Anticalin fusions
The restriction endonuclease FokI from Flavobacterium okeanokoites is a well studied protein. It consists of two domains, a DNA recognition domain and a DNA cleavage domain. Upon recognition of one target site and dimerization it cleaves the DNA nine bases apart from the recognition site. Several groups reported experiments to uncouple the cleavage and restriction domains of FokI and created a novel site-specific endonucleases by linking the cleavage domain to zinc finger proteins (Miller et al. 2007).
For our project we combined two previous research results and generated a Fok cleavage heterodimer comprising an enzymatically active and inactive monomer. For the catalytic active Fok partner, named Fok_a, as well as for the catalytic inactive Fok partner, Fok_i, the association interface was mutated to disfavor homodimerization and promote heterodimerization. In Fok_i additional amino acid exchanges led to the inactivation.
The two heterodimeric partners were fused to anticalins binding different adapter molecules. Fok_a is genetically fused to a digoxigenin-binding anticalin (DigA) and Fok_i to a fluorescein-binding anticalin (FluA). The adapter molecules digoxigenin and fluorescein are common modifications linked to oligonucleotides thus mediating the binding to the DNA site of interest. Two modifications allow for a better spatial control of the cleavage site. On the target site Fok_i and Fok_a constructs are brought into close proximity and can form a heterodimer. The inactive Fok domain will serve as an activator of the active Fok domain, which cuts one strand of the DNA. Our structural ‘3D’ models, indicate that Fok domains can be positioned in such a way that Fok_a will cleave the target DNA, and Fok_i would be directed towards the modified oligonucleotide. Different linkers were designed and fused between cleavage domain and the anticalin binding moieties to test for the optimal distance. In addition we generated a monomeric Fok fusion construct, to enable phage display and te4st a further setting requiring only one binding domain and hapten. Furthermore, we made a Fok cleavage domain fusion with a coiled coil based DNA binding domain, because we can combine these with existing light switchable inhibitors, which prevent DNA binding. As a result we will obtaina light switchable restriction enzyme.
Milestones
To reach our goal within the short given time frame we started several subprojects in parallel. Our subprojects listed here are defined along these projects.
Designing of the constructs was aided by extensive model building and analysis of the spatial orientation of the different proteins and oligonucleotides used. All designed and constructed parts feature full BioBrick compatibility and in addition allow for the construction of fusion proteins based on the RFC 25 (Freiburg) cloning standard. Cloning of the respective parts was followed by expression purification and analysis. Despite previous literature data our active Fok construct was toxic to cells when expressed and we tested periplasmic expression with export of the nascent polypeptide chain before folding.
In our experiments we addressed the following questions:
• Structural Model building
• Design of protein fusion parts
• Cloning of anticalin Fok fusions
• Cloning of a monomeric Fok construct and of a Jun/Fos directed Fok construct
• Expression and purification of constructs
• In vitro assays
• In vivo assays
• Phage Display of an Ago protein
• Modeling of assembly and cleavage with differential equations
• An international survey of laymen on synthetic biology
In short, we successfully worked on all aspects. Experiments to validate our approach in more compelx settings are ongoing.
The labs of Kristian Müller and Katja Arndt provided all technology and support for the project.
 Schematic Model of the universal restriction enzymes based on FokI and anticalins. |
 Structural model of the universal restriction enzymes based on FokI and anticalins. |
 Model of the catalytic cycle; hybridization - cleavage - temperature promoted release. |
 Structure of an Ago protein, demonstrating guide oligonucleotide mediated binding. |
Modeling of the Enzyme Kinetics
| For our modeling analyses we constructed various sets of differential equations describing
protein-protein and protein-DNA interactions and the final cleavage. Read more... |
Modeling of the Enzyme Structure
| Structural modeling was an initial step towards our planned universal restriction enzyme. Using molecular display software we arranged published crystal structures of FokI, anticalins and DNA. The arrangement was guided by superimposition with further structures of enzyme bound DNA. The modeling defined spatial requirements for linker lengths and positioning of modifications within oligonucleotides. Read more... |  |
In vitro assays
| After the cloning, expression and the purification of the Fok constructs we conducted several assays in order to to analyze the activity of the enzyme. To establish the assay and as a reference for activity we used wild type FokI. Binding of the modified nucleotides and enzymatic activity were tested with the Fok_i / Fok_a construct. Read more... |  |
 |
In vivo Assays

|
We demonstrated that modified guide oligonucleotides can be transfected in E. coli. Importantly, we showed that a M13 DNA hybridized with a guide oligonucleotide does not produce phages when transfectd in cells expressing Fok fusion proteins whereas the control M13 DNA does. Read more... |
Protein Expression and Purification
| Proteins were recombinantly expressed in E. coli strains BL21 or RV308. Purification was achieved by affinity chromatography utilizing the His-tag, Strep-tag, or the GST fusion. If needed, two affinity purifications due to two available tags were combined or a size exclusion chromatogrpahy was added. Read more... | .JPG)
|
AGO
| The Argonaute proteins represent our second approach to generate universal restriction enzymes. These RNases provide already important features such as recognition of guide oligonucleotides and thermostability. We expressed and purified an AGO protein and showed residual DNase activtiy. Consequently, we started phage display of error prone PCR diversified AGO in order to convert the RNase in a DNase. We demonstratd AGO display, successfully completed two rounds of phage display and identified interesting mutants. Read more... |  |
Ethics
| We conducted an international survey on benefit and risk perception of Synthetic Biology by laymen, which was translated in 10 languages. Read more... |
Fok Monomer
 |
To ease production, purification, handling and analysis we cloned a monomeric Fok variant. In addition, this variant is better suited for phage display, which will allow us selection for improved properties. This protein comprises four functional subunits interspaced by two short and one long linker (anticalin-Fok_i-long_linker-Fok_a-anticalin). Read more... |
Alternative way of binding: Jun/Fos
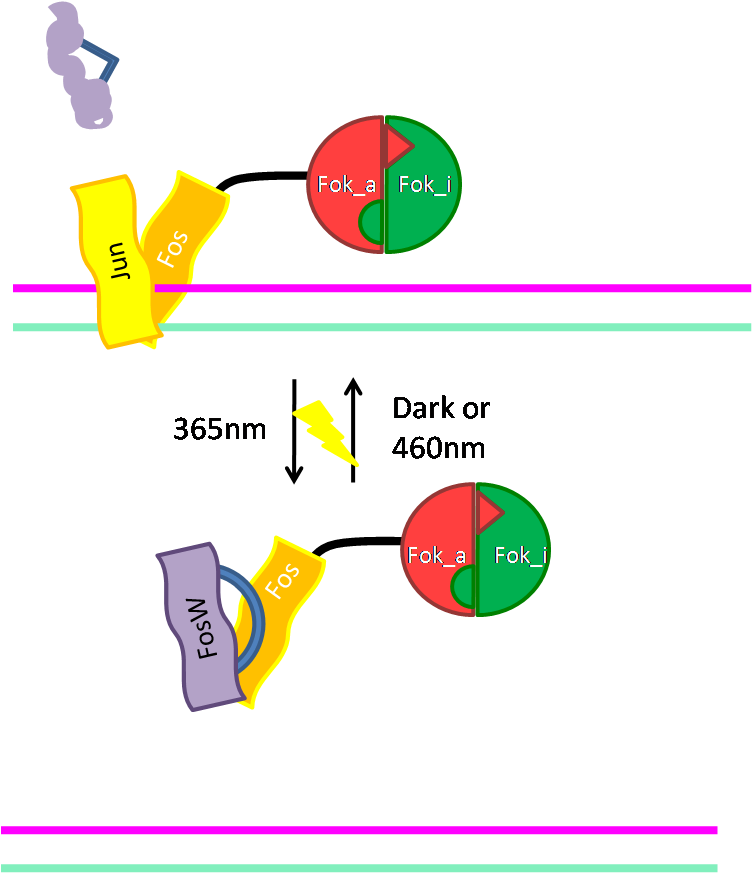 |
As an alternative way of binding of Fok to DNA we cloned the DNA binding domain (bZIP) of the activator protein-1 (AP-1). As light switching of coiled coils is established in our host lab, this will give us a light switchable restriction enzyme. Read more... |
Cloning strategy
| Here we give an overview of the cloning strategies we used for creating a universal restriction endonuclease. Fusion proteins were generated according to the Freiburg Assembly standard RFC 25. Read more... |
contact: freigem09@googlemail.com; kristian@biologie.uni-freiburg.de