 "
"
Team:IIT Madras/Project
From 2009.igem.org
Ramakrishna (Talk | contribs) (→Circuit) |
(→Theory) |
||
| Line 187: | Line 187: | ||
Second, it was experimentally found that plasmid-bearing cells usually have a lower maximum specific growth rate than their plasmid-free counterparts, and once a plasmid-free cell arises, it competes with its plasmid-bearing counterparts very rapidly and ultimately phases it out. Since most recombinant plasmids are not conjugative (not capable of self-transfer to other plasmid-free cells), if a cell has lost a plasmid, there is no way for the cell to acquire it again. Thus, a segregation of plasmids at cell division and the difference in the growth rates of plasmid-free and plasmid bearing subpopulations determine the rate at which plasmids are lost during prolonged cultivation. Here, we use plasmids which can confer resistance to certain antibiotics in the medium and link them up in a certain way so that they repress the expression of the gene of our interest. As the selection pressure is removed, the plasmids which have the repressors for the gene of interest are lost, hence revealing the gene on using the correct series of antibiotic washes. | Second, it was experimentally found that plasmid-bearing cells usually have a lower maximum specific growth rate than their plasmid-free counterparts, and once a plasmid-free cell arises, it competes with its plasmid-bearing counterparts very rapidly and ultimately phases it out. Since most recombinant plasmids are not conjugative (not capable of self-transfer to other plasmid-free cells), if a cell has lost a plasmid, there is no way for the cell to acquire it again. Thus, a segregation of plasmids at cell division and the difference in the growth rates of plasmid-free and plasmid bearing subpopulations determine the rate at which plasmids are lost during prolonged cultivation. Here, we use plasmids which can confer resistance to certain antibiotics in the medium and link them up in a certain way so that they repress the expression of the gene of our interest. As the selection pressure is removed, the plasmids which have the repressors for the gene of interest are lost, hence revealing the gene on using the correct series of antibiotic washes. | ||
| - | + | '''Fig 3''':[[Image:expressionofgeneofinterest.jpg|500px]] | |
=== Circuit === | === Circuit === | ||
Revision as of 10:25, 18 October 2009
IIT Madras
Our project PLASMID - Plasmid Locking Assembly for Sustaining Multiple Inserted DNA - introduces a new paradigm in gene regulation. The study is based on the concept of plasmid loss. Any episome introduced into the cell shows a segregational asymmetry accompanied with differential growth rates in the absence and presence of episome leading to an overall loss of the episomal unit in the absence of any maintaining selective pressure. It is hypothesized that by appropriately controlling the external selective pressures, one can control the direction of plasmid loss in the cell, modifying the existing gene regulation system in a pre determined manner. It is also hypothesized that introducing negative selective pressures against certain other directions of plasmid loss, in the form of constitutively repressed endotoxins will help streamline the regulatory system even further.
If successful, this study allows for exquisitely delicate and precise multifactorial control of gene regulation in the future. This model can also be used to hide genes of commercial interest to protect it from unauthorized use (under some conditions).
Contents |
Summary
Our project is based on the fundamental concept of plasmid instability in a novel way to conceal information or ‘lock’ a gene’s function in a cell until the correct combination of inputs is fed into the cell. We call this a ‘combinatorial lock’ or PLASMID. It involves the positive regulation of the gene of interest only on receiving the correct inputs from the user. We use plasmids which can confer resistance to certain antibiotics in the medium and link them up in a certain way (i.e, essentially designing a genetic circuit) so that they repress the expression of the gene of our interest. As the selection pressure is lifted from the media, the plasmids which have the repressors for the gene of interest are lost, hence revealing the gene on using the correct series of antibiotic washes. In essence, the process of unlocking would simply be the correct sequence of antibiotic media in which the cells should be washed. We would be working with a 2 plasmid system and it is easy to see that this principle, theoretically, could be extended to N plasmids. In general the code length required to "unlock" is N-1 if the number of plasmids introduced are N. In our case, since the number of plasmids being introduced are 2, the code would essentially be just 1 unit long. Particularly in this case, the 1 unit of code corresponds to growing the cells in one correct antibiotic medium.
Fig1: picture..picture...picture...picture..picture (of a network of plasmids and a gene of interest being represed)
However, in the experiments we have not incorporated any particular gene to be repressed. instead we study how can we achieve a directed loss of plasmids which is the idea central to the working of the system. In place of a gene of interest, we have placed fluorescent reporters in each plasmid to monitor the presence or absence of any particular palsmid.
Theory
The most important idea behind the working of the lock is plasmid loss due to lack of selection. Any extra-chromosomal genetic material introduced into the cell tends to disappear over the generations, unless they confer a selective survival advantage over the cells that do not possess the plasmid. During the growth of bacteria, plasmid-free variants arise in the initially homogeneous plasmid-bearing cell population basically in two ways. First, each plasmid-bearing cell has a certain probability to give rise to a plasmid-free cell at cell division (this depends on the mechanisms of plasmid distribution between daughter cells, plasmid copy number at the cell division, the presence of multimer resolution loci, etc.).
Fig 2.1: Fig 2.2:
Fig 2.2:
Usually, such probability is very low for natural plasmids (about 10-7) whereas recombinant plasmids (i.e. genetically modified) may segregate with a higher probability (10-3—10-5). Several hypotheses have been put forward to explain this tremendous difference: impaired copy number control, the absence/impairment of the multimer resolution genes and random distribution of plasmid among daughter cells for low copy number plasmids. Second, it was experimentally found that plasmid-bearing cells usually have a lower maximum specific growth rate than their plasmid-free counterparts, and once a plasmid-free cell arises, it competes with its plasmid-bearing counterparts very rapidly and ultimately phases it out. Since most recombinant plasmids are not conjugative (not capable of self-transfer to other plasmid-free cells), if a cell has lost a plasmid, there is no way for the cell to acquire it again. Thus, a segregation of plasmids at cell division and the difference in the growth rates of plasmid-free and plasmid bearing subpopulations determine the rate at which plasmids are lost during prolonged cultivation. Here, we use plasmids which can confer resistance to certain antibiotics in the medium and link them up in a certain way so that they repress the expression of the gene of our interest. As the selection pressure is removed, the plasmids which have the repressors for the gene of interest are lost, hence revealing the gene on using the correct series of antibiotic washes.
Fig 3: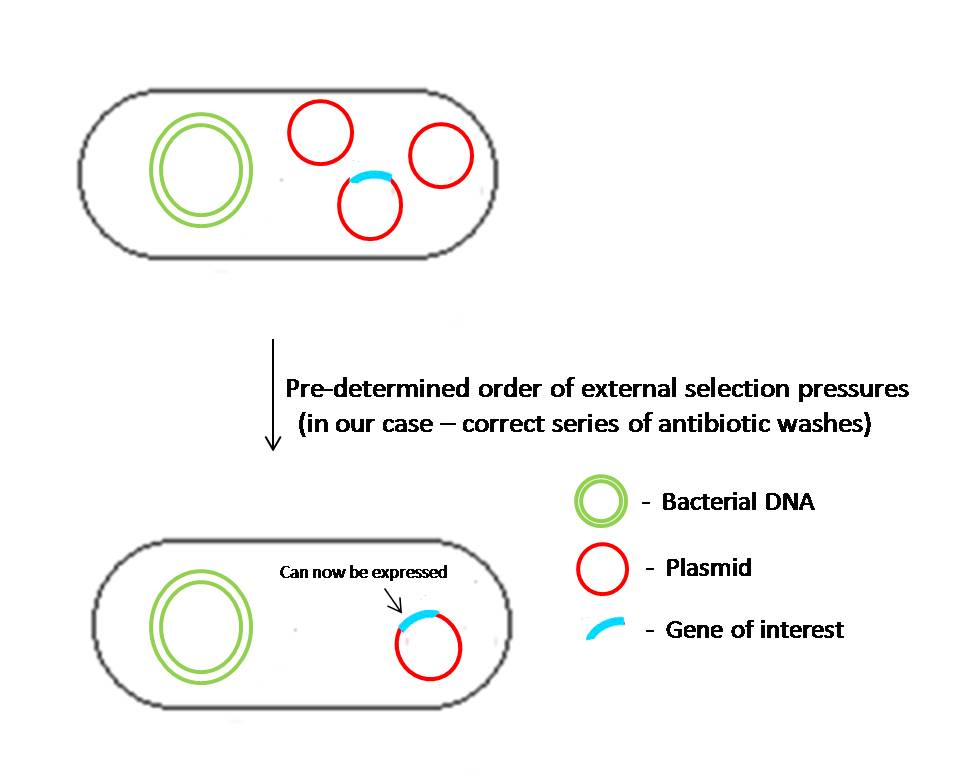

Circuit
One of the plasmids will have the gene of interest which will be expressed only when the plasmids apart from this particular one are lost in a particular order. This particular order or “code” will consist of a sequence of antibiotic treatments given to the transformed cells. The correct code triggers the loss of plasmids in the cells in a particular order. Each plasmid will be linked to the plasmids which are supposed to be lost before and after it in a very tightly regulated fashion. The regulation of the plasmid loss can be regulated very tightly using a “suicide gene” (gene coding for the bacterial gyrase poison). These genes will be triggered when the culture is subjected to the wrong antibiotic (Out of sequence). To ensure that the suicide genes don’t fire randomly, they are under the control of repressors which are on the plasmids that are supposed to be lost after it and are expressed constitutively. Thus, they fire only when the plasmid containing the repressor is lost. When all the plasmids other than the plasmid containing the gene of interest are lost, the repressors that have been blocking the required gene are lost, thus allowing its expression, which can manifest as a phenotype or a function in the cell. This will be the “unlocking” of the lock.
The grand idea of a 3 plasmid locking system revolved around constructing something like the below
Fig3.1: 
To which we thought we could fit in the following parts

Then we scaled it down and began to build constructs for a 2 plasmid system which works like this
Fig 3.2: pic pic pic 
To which we fit in parts from the registry to make it look like this

For the proof of concept experiments, we built the following constructs
[http://partsregistry.org/Part:BBa_K272001 K272001-constitutive RFP expressor]

[http://partsregistry.org/Part:BBa_K272002 K272002-constitutive CFP expressor]

Expected behavior of the system
Fig4: picture..picture...picture...picture..picture (2 plasmid case)
Fig5: picture..picture...picture...picture..picture
Work Plan
Building the constructs
We have been following 3 different strategies to build our constructs. 1) The 3A assembly
2) The 2A or Standard Assembly 3) PCR based approach: We plan to proceed with the experiment using a PCR based amplification system, followed by a specific restriction digest and subsequent ligation.
working with the construct
We will be performing a proof-of-concept experiment using the 2 plasmids - K272001 and K272002. The cells will be co-transformed with the plasmids. Each of these plasmids have a different fluorescent reporter and their backbones have different antibiotic resistances. However, the origins of replication in both the plasmids are the same, thus creating a tight competition between the plasmids to remain in the cell based on the selection pressures. The bacteria "titrates" the number of plasmids with a certain origin of replication before each replication. Thus if there are a couple of plasmids with the same origin of replication, and one of them happens to be non essential and does not confer any advantage to the cell for a given growth condition it will eventually be phased out from the population. This is essentially mimicking natural selection. The fluorescent reporters will help in tracing which plasmid is phasing out for a given set of growth conditions.
The cotransformed cells will be grown in different Antibiotic media and the loss of each Plasmid will be studied using fluorescent markers under a microscope. Presently we are working with the K272001 (constitutive RFP) in pSB1C3 and K272002 (constitutive CFP) in pSB1A2. We expect to see that the cotransformed cells when grown in a medium with both chloramphenicol and ampicillin should have more number of cells with both the fluorescent reporters than with those which are grown in medium containing either or none of the antibiotics. Specifically, if the cells are grown in medium containing only ampicillin, then the number of cells without CFP should increase and when grown in the absence of chloramphenicol, the number of cells without RFP should be on the rise.
pic..pic...pic..pic..pic...
The constructs required to demonstrate the locking property are being built and will be tested once the construction is done.