 "
"
Team:Todai-Tokyo/Notebook/isoleucine
From 2009.igem.org
(→August) |
(changed "zenaku" characters to "hankaku") |
||
| Line 39: | Line 39: | ||
*Mix 1ul of DNA with 100ul of competent cells on ice. | *Mix 1ul of DNA with 100ul of competent cells on ice. | ||
*Leave on ice for 30 minutes. | *Leave on ice for 30 minutes. | ||
| - | *Heat shock at | + | *Heat shock at 42°Cfor 45 seconds. |
*Leave on ice for 2 minutes. | *Leave on ice for 2 minutes. | ||
| - | *Add 500ul of LB and incubate at | + | *Add 500ul of LB and incubate at 37°C for 1 hour. |
*Plate on LB-ampicillin plates. | *Plate on LB-ampicillin plates. | ||
| Line 93: | Line 93: | ||
Performed PCR using the following program: | Performed PCR using the following program: | ||
| - | 1. | + | 1. 95°C 2 minutes<Br> |
| - | 2. | + | 2. 95°C 30 seconds<Br> |
| - | 3. | + | 3. 50°C 30 seconds<Br> |
| - | 4. | + | 4. 72°C 40 seconds<Br> |
5. Repeat 2-4 23 times<Br> | 5. Repeat 2-4 23 times<Br> | ||
| - | 6. | + | 6. 25°C forever<Br> |
Purified PCR product using the Promega PCR purification kit. | Purified PCR product using the Promega PCR purification kit. | ||
| Line 126: | Line 126: | ||
10ul MilliQ<Br> | 10ul MilliQ<Br> | ||
| - | Incubated mixtures at | + | Incubated mixtures at 37°C for 1 hour. |
Ran on gel to gel purify:<BR> | Ran on gel to gel purify:<BR> | ||
| Line 202: | Line 202: | ||
| - | 1. | + | 1. 95°C 2 minutes |
| - | 2. | + | 2. 95°C 30 seconds |
| - | 3. | + | 3. 50°C 30 seconds |
| - | 4. | + | 4. 72°C 40 seconds |
5. Repeat 2-4 29 times | 5. Repeat 2-4 29 times | ||
| - | 6. | + | 6. 25°C forever |
| Line 242: | Line 242: | ||
| - | Incubated mixtures at | + | Incubated mixtures at 37°C for 10 minutes. |
| Line 291: | Line 291: | ||
Performed PCR using the following program: | Performed PCR using the following program: | ||
| - | 1. | + | 1. 95°C 2 minutes<Br> |
| - | 2. | + | 2. 95°C 30 seconds<Br> |
| - | 3. | + | 3. 50°C 30 seconds<Br> |
| - | 4. | + | 4. 72°C 40 seconds<Br> |
5. Repeat 2-4 29 times<Br> | 5. Repeat 2-4 29 times<Br> | ||
| - | 6. | + | 6. 25°C forever<Br> |
Purified PCR product using the Promega PCR purification kit. | Purified PCR product using the Promega PCR purification kit. | ||
| Line 326: | Line 326: | ||
28ul MilliQ<Br> | 28ul MilliQ<Br> | ||
| - | Incubated mixtures at | + | Incubated mixtures at 37°C for 1 hour. |
Ran on gel to gel purify:<BR> | Ran on gel to gel purify:<BR> | ||
| Line 345: | Line 345: | ||
TaKaRa Solutions 1 4ul<BR> | TaKaRa Solutions 1 4ul<BR> | ||
| - | Incubated mixtures at | + | Incubated mixtures at 37°C for 10 minutes. |
'''Transformation'''<BR> | '''Transformation'''<BR> | ||
| Line 362: | Line 362: | ||
Performed PCR using the following program: | Performed PCR using the following program: | ||
| - | 1. | + | 1. 94°C 2 minutes<Br> |
| - | 2. | + | 2. 94°C 30 seconds<Br> |
| - | 3. | + | 3. 52°C 80 seconds<Br> |
| - | 4. | + | 4. 72°C 60 seconds<Br> |
5. Repeat 2-4 29 times<Br> | 5. Repeat 2-4 29 times<Br> | ||
| - | 6. | + | 6. 25°C forever<Br> |
lane1:marker<BR> | lane1:marker<BR> | ||
| Line 376: | Line 376: | ||
PCR successful!<BR> | PCR successful!<BR> | ||
But we couldn't do ligation because TA cloning kit was not left.<BR> | But we couldn't do ligation because TA cloning kit was not left.<BR> | ||
| - | We purified PCR products and put them in - | + | We purified PCR products and put them in -20°C. |
| Line 383: | Line 383: | ||
put a small amount of single colony into each tube with 5ul MilliQ water<BR> | put a small amount of single colony into each tube with 5ul MilliQ water<BR> | ||
| - | + | ↓<BR> | |
| - | + | 95°C 5min<BR> | |
| - | + | ↓<BR> | |
PCR reaction<BR> | PCR reaction<BR> | ||
1ul 10×buffer <BR> | 1ul 10×buffer <BR> | ||
| Line 393: | Line 393: | ||
0.1ul 3’-primer<BR> | 0.1ul 3’-primer<BR> | ||
2.92ul MilliQ water<BR> | 2.92ul MilliQ water<BR> | ||
| - | + | ↓<BR> | |
added the PCR reaction to each tube.<BR> | added the PCR reaction to each tube.<BR> | ||
| - | + | ↓<BR> | |
Performed PCR using the following program:<BR> | Performed PCR using the following program:<BR> | ||
| - | 1. | + | 1. 95°C 2min<BR> |
| - | 2. | + | 2. 95°C 30sec <BR> |
| - | 3. | + | 3. 52°C 30sec<BR> |
| - | 4. 72. | + | 4. 72.5°C 80sec<BR> |
5. repeat 2-4 29times<BR> | 5. repeat 2-4 29times<BR> | ||
| - | 6. | + | 6. 25°C forever<BR> |
PCR successful!<BR> | PCR successful!<BR> | ||
| Line 419: | Line 419: | ||
1ul plasmid(0.15ug/ul)<BR> | 1ul plasmid(0.15ug/ul)<BR> | ||
0.5ul 5'or3'primer(3.2pmol/ul)<BR> | 0.5ul 5'or3'primer(3.2pmol/ul)<BR> | ||
| - | + | ↓<BR> | |
PCR Program<BR> | PCR Program<BR> | ||
| - | 1. | + | 1.96°C 2min<BR> |
| - | 2. | + | 2.96°C 10sec<BR> |
| - | 3. | + | 3.55°C 5sec<BR> |
| - | 4. | + | 4.60°C 3min <BR> |
5.go to 2.29times<BR> | 5.go to 2.29times<BR> | ||
| - | 6. | + | 6.25°C forever<BR> |
| - | + | ↓<BR> | |
add 0.5ul PHOSPHATASE ALKALINE shrimp<BR> | add 0.5ul PHOSPHATASE ALKALINE shrimp<BR> | ||
| - | + | ↓<BR> | |
| - | + | 37°C 1hr incubate<BR> | |
| - | + | ↓<BR> | |
add 1ul 3MNaOAc<BR> | add 1ul 3MNaOAc<BR> | ||
| - | + | ↓<BR> | |
add 25ul EtOH<BR> | add 25ul EtOH<BR> | ||
| - | + | ↓<BR> | |
| - | 20000xg | + | 20000xg 4°C 10min centrifugation<BR> |
| - | + | ↓<BR> | |
put off supernatant<BR> | put off supernatant<BR> | ||
| - | + | ↓<BR> | |
dry tubes<BR> | dry tubes<BR> | ||
| - | + | ↓<BR> | |
add 15ul HiDi<BR> | add 15ul HiDi<BR> | ||
| - | + | ↓<BR> | |
put them in the sequence machine<BR> | put them in the sequence machine<BR> | ||
| Line 474: | Line 474: | ||
Performed PCR using the following program: | Performed PCR using the following program: | ||
| - | 1. | + | 1. 95°C 2 minutes<Br> |
| - | 2. | + | 2. 95°C 30 seconds<Br> |
| - | 3. | + | 3. 55°C 30 seconds<Br> |
| - | 4. 72. | + | 4. 72.5°C 30 seconds<Br> |
5. Repeat 2-4 29 times<Br> | 5. Repeat 2-4 29 times<Br> | ||
| - | 6. | + | 6. 25°C forever<Br> |
<BR> | <BR> | ||
restriction enzyme fragmentation of double terminator by XbaI<BR> | restriction enzyme fragmentation of double terminator by XbaI<BR> | ||
| Line 487: | Line 487: | ||
2.5ul XbaI<BR> | 2.5ul XbaI<BR> | ||
up to 100ul MilliQ water<BR> | up to 100ul MilliQ water<BR> | ||
| - | + | 37°C over night<BR> | |
restriction enzyme fragmentation of double terminator by EcoRI<BR> | restriction enzyme fragmentation of double terminator by EcoRI<BR> | ||
1ug plasmid<BR> | 1ug plasmid<BR> | ||
| Line 493: | Line 493: | ||
2.5ul EcoRI<BR> | 2.5ul EcoRI<BR> | ||
up to 100ul MilliQ water<BR> | up to 100ul MilliQ water<BR> | ||
| - | + | 37°C over night<BR> | |
restriction enzyme fragmentation of YqiT gene by SpeI<BR> | restriction enzyme fragmentation of YqiT gene by SpeI<BR> | ||
1ug DNA<BR> | 1ug DNA<BR> | ||
| Line 499: | Line 499: | ||
2.5ul SpeI<BR> | 2.5ul SpeI<BR> | ||
up to 100ul MilliQ water<BR> | up to 100ul MilliQ water<BR> | ||
| - | + | 37°C over night<BR> | |
restriction enzyme fragmentation of YqiT gene by EcoRI<BR> | restriction enzyme fragmentation of YqiT gene by EcoRI<BR> | ||
1ug DNA<BR> | 1ug DNA<BR> | ||
| Line 505: | Line 505: | ||
2.5ul EcoRI<BR> | 2.5ul EcoRI<BR> | ||
up to 100ul MilliQ water<BR> | up to 100ul MilliQ water<BR> | ||
| - | + | 37°C over night<BR> | |
=='''8/15,16'''== | =='''8/15,16'''== | ||
| Line 519: | Line 519: | ||
Performed PCR using the following program:<BR> | Performed PCR using the following program:<BR> | ||
| - | 1. | + | 1. 95°C 2 minutes<BR> |
| - | 2. | + | 2. 95°C 30 seconds<BR> |
| - | 3. | + | 3. 55°C 30 seconds<BR> |
| - | 4. 72. | + | 4. 72.5°C 30 seconds<BR> |
5. Repeat 2-4 29 times<BR> | 5. Repeat 2-4 29 times<BR> | ||
| - | 6. | + | 6. 25°C forever<BR> |
*cut YqiT off with EcoRI and SpeI<BR> | *cut YqiT off with EcoRI and SpeI<BR> | ||
| Line 542: | Line 542: | ||
*PCR of Ahr and Arnt for TA cloning(Ex-taq , Funakoshi)<BR> | *PCR of Ahr and Arnt for TA cloning(Ex-taq , Funakoshi)<BR> | ||
template:human Ahr cDNA and human Arnt cDNA<BR> | template:human Ahr cDNA and human Arnt cDNA<BR> | ||
| - | 1, | + | 1,95°C 2min<BR> |
| - | 2, | + | 2,95°C 30sec<BR> |
| - | 3, | + | 3,55°C 30sec<BR> |
| - | 4,72. | + | 4,72.5°C 1.5min<BR> |
| - | 5, | + | 5,go°C to 2, 29times<BR> |
| - | 6, | + | 6,25°C forever<BR> |
<BR> | <BR> | ||
<BR> | <BR> | ||
| - | =='''October'''= | + | =='''October'''='em |
*PCR of gal1/10 promoter (Ex-taq , Funakoshi)<BR> | *PCR of gal1/10 promoter (Ex-taq , Funakoshi)<BR> | ||
template:S.serevisiae genome DNA<BR> | template:S.serevisiae genome DNA<BR> | ||
| - | 1, | + | 1,95°C 2min<BR> |
| - | 2, | + | 2,95°C 30sec<BR> |
| - | 3, | + | 3,55°C 30sec<BR> |
| - | 4,72. | + | 4,72.5°C1min<BR> |
| - | 5, | + | 5,go°C to 2, 29times<BR> |
| - | 6, | + | 6,25°C forever<BR> |
<BR> | <BR> | ||
*TA cloning of Ahr, Arnt and gal1/10 promoter<BR> | *TA cloning of Ahr, Arnt and gal1/10 promoter<BR> | ||
ligate PCR product and T bector<BR> | ligate PCR product and T bector<BR> | ||
| - | + | ↓<BR> | |
put each plasmid into different E.coli<BR> | put each plasmid into different E.coli<BR> | ||
| - | + | ↓<BR> | |
Miniprep of them by using Promega kit<BR> | Miniprep of them by using Promega kit<BR> | ||
| - | + | ↓<BR> | |
read each sequence in order to check whether there is plasmids including Ahr, Arnt or gal1/10 promoter<BR> | read each sequence in order to check whether there is plasmids including Ahr, Arnt or gal1/10 promoter<BR> | ||
<BR> | <BR> | ||
| Line 585: | Line 585: | ||
cut YqiT-double terminator off with XbaI and PstI<BR> | cut YqiT-double terminator off with XbaI and PstI<BR> | ||
cut P2-10F, P2-16E, P1-22A and P1-21B off with SpeI and PstI<BR> | cut P2-10F, P2-16E, P1-22A and P1-21B off with SpeI and PstI<BR> | ||
| - | + | ↓<BR> | |
ligate YqiT-double terminator and iGEM part<BR> | ligate YqiT-double terminator and iGEM part<BR> | ||
| - | + | ↓<BR> | |
put each plasmid into different E.coli<BR> | put each plasmid into different E.coli<BR> | ||
| - | + | ↓<BR> | |
Miniprep of them by using Promega kit<BR> | Miniprep of them by using Promega kit<BR> | ||
| - | + | ↓<BR> | |
read each sequence in order to check whether there is plasmids including the construct<BR> | read each sequence in order to check whether there is plasmids including the construct<BR> | ||
<BR> | <BR> | ||
Revision as of 16:23, 18 October 2009

| Home | The Team | The Project | Parts Submitted to the Registry | Modeling | Notebook | Protocols | Ethics |
|---|
Contents |
Plan
Aim: Create bacteria that produce a noxious odour when induced to do so
Methods:
- Clone the yqiT gene from Bacillus subtilis into a biobrick vector.
- Make constructs that express this gene constitutively and under the control of an inducible promoter.
6/5
Constructs to be created:
- yqiT
- ptetR-RBS-yqiT-dterm
- pAraC-RBS-yqiT-dterm
- pLacI-RBS-yqiT-dterm
Obtaining DNA:
Resuspended DNA in the following wells with 10ul water:
Plate 1 1D
[http://partsregistry.org/wiki/index.php/Part:BBa_R0080 AraC regulated promoter]
Plate 1 12E
[http://partsregistry.org/wiki/index.php?title=Part:BBa_R0010 LacI regulated promoter]
Plate 1 1H
[http://partsregistry.org/wiki/index.php/Part:BBa_B0030 Strong RBS]
Plate 1 13B
[http://partsregistry.org/wiki/index.php?title=Part:BBa_J13002 TetR repressed POPS generator]
Plate 2 24C
[http://partsregistry.org/wiki/index.php?title=Part:BBa_B0014 Double terminator]
Transformed 1ul of each of the above into DH5a competent cells:
Transformation
- Mix 1ul of DNA with 100ul of competent cells on ice.
- Leave on ice for 30 minutes.
- Heat shock at 42°Cfor 45 seconds.
- Leave on ice for 2 minutes.
- Add 500ul of LB and incubate at 37°C for 1 hour.
- Plate on LB-ampicillin plates.
6/6
No colonies grew from the 5 transformations of 6/5.
6/7
Creating a biobrick part out of yqiT
Strategy: PCR out the yqiT gene from the Bacillus subtilis genome using primers to attach the biobrick preffix/suffix and clone this into a biobrick vector. The vector utilized is the GFP generator which houses a sufficient-sized insert:
Plate 1 16E (from 2007 iGEM distribution provided by Chiba University)
[http://partsregistry.org/wiki/index.php?title=Part:BBa_E0840 GFP generator]
Overview:
- PCR yqiT gene from genome
- purify DNA from the PCR product
- digest GFP generator and purified PCR product by XbaI/PstI
- gel-purify the digested vector (GFP generator without the insert i.e. empty) and the yqiT gene
- ligate the above 2 fragments of DNA together
- transform into E. coli and select for ampicillin resistance
- check for transformation success using colony PCR by yqiT primers
PCR of yqiT gene
The Bacillus subtilis genome was provided by Bacillus subtilis 168
The following primers were used to amplify an approx. 1500bp fragment from the Bacillus subtilis genome.
yqiT EX: ccggaattctctagaatggaactttttaaatatatggaacgatacg(bold text is a sequence binding yqiT gene)
yqiT SP: ctgcagcggccgctactagtattagcgacgacttaaaatatgttgg(bold text is a sequence binding yqiT gene)
PCR protocol
The following was mixed in a PCR tube:
1ul 20uM yqiT EX primer
1ul 20uM yqiT SP primer
2ul 10X Pfu Ultra buffer
1.6ul dNTP mix
0.15ul Bacillus subtilis genome
0.5ul Pfu Ultra enzyme
13.85ul MilliQ
Performed PCR using the following program:
1. 95°C 2 minutes
2. 95°C 30 seconds
3. 50°C 30 seconds
4. 72°C 40 seconds
5. Repeat 2-4 23 times
6. 25°C forever
Purified PCR product using the Promega PCR purification kit.
Ran on gel to visualize bands:

PCR unsuccessful: will digest the GFP generator but cannot perform the ligation without the PCR product.
Digestion
Digested purified PCR product and the GFP generator both with XbaI/PstI:
yqiT
3ul PCR product
1ul High buffer
0.5ul XbaI
0.5ul PstI
5ul MilliQ
GFP generator
6ul DNA
2ul High buffer
1ul XbaI
1ul PstI
10ul MilliQ
Incubated mixtures at 37°C for 1 hour.
Ran on gel to gel purify:
Lanes 1, 12:marker
Lanes 2, 5, 9:GFP
Lanes 3, 6, 10:yqiT

Cut out the second band from the bottom and dissolved the gel using the Promega gel purification kit to extract DNA from.
This DNA is stored for later usage as the GFP generator X/P vector fragment.
6/14
Creating a biobrick part out of yqiT
Strategy: PCR out the yqiT gene from the Bacillus subtilis genome using primers to attach the biobrick preffix/suffix and clone this into a biobrick vector. The vector utilized is the GFP generator which houses a sufficient-sized insert:
Plate 1 1D (from 2009 iGEM distribution provided by Tokyo University)
promoter (lacI regulated)
Overview:
PCR yqiT gene from genome
gel-purify DNA from the PCR product
digest lacI regulated promoter and purified PCR product by XbaI/PstI
gel-purify the digested vector (lacI regulated promoter without the insert i.e. empty) and the yqiT gene
ligate the above 2 fragments of DNA together
transform into E. coli and select for ampicillin resistance
check for transformation success using colony PCR by yqiT primers
PCR of yqiT gene The Bacillus subtilis genome was provided by Bacillus subtilis 168
The following primers were used to amplify an approx. 1095bp fragment from the Bacillus subtilis genome.
yqiT EX: ccggaattctctagaatggaactttttaaatatatggaacgatacg(bold text is a sequence binding yqiT gene)
yqiT SP: ctgcagcggccgctactagtattagcgacgacttaaaatatgttgg(bold text is a sequence binding yqiT gene)
PCR protocol
The following was mixed in a PCR tube:
1ul 20uM yqiT EX primer
1ul 20uM yqiT SP primer
2ul 10X Pfu Ultra buffer
1.6ul dNTP mix
0.15ul Bacillus subtilis genome
0.5ul Pfu Ultra enzyme
13.75ul MilliQ
Performed PCR using the following program:
1. 95°C 2 minutes
2. 95°C 30 seconds
3. 50°C 30 seconds
4. 72°C 40 seconds
5. Repeat 2-4 29 times
6. 25°C forever
Purified PCR product using the Promega PCR purification kit.
Ran on gel to gel purify:
Lanes 1:marker 6ul
Lanes 2:yqiT PCR product 10ul + 10xLoading Dye 2ul
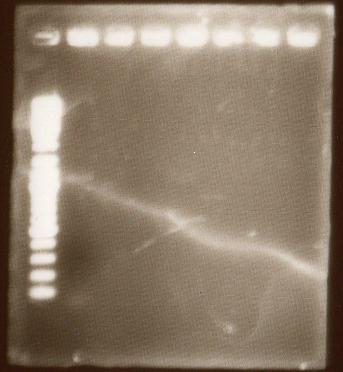

PCR successful
Ligation
Ligated gel purified yqiT and vector with TaKaRa Solutions 1:
lacI regulated promoter (vector) 2ul
yqiT 8ul
TaKaRa Solutions 1 6ul
Incubated mixtures at 37°C for 10 minutes.
Transformation
6/21
Creating a biobrick part out of yqiT
Strategy: PCR out the yqiT gene from the Bacillus subtilis genome using primers to attach the biobrick preffix/suffix and clone this into a biobrick vector. The vector utilized is the GFP generator which houses a sufficient-sized insert:
Plate 1 1D (from 2009 iGEM distribution provided by Tokyo University)
[http://partsregistry.org/wiki/index.php?title=Part:BBa_R0010 promoter (lacI regulated)]
Overview:
- PCR yqiT gene from genome
- gel-purify DNA from the PCR product
- digest lacI regulated promoter and purified PCR product by XbaI/PstI
- gel-purify the digested vector (lacI regulated promoter without the insert i.e. empty) and the yqiT gene
- ligate the above 2 fragments of DNA together
- transform into E. coli and select for ampicillin resistance
- check for transformation success using colony PCR by yqiT primers
PCR of yqiT gene
The Bacillus subtilis genome was provided by Bacillus subtilis 168
The following primers were used to amplify an approx. 1095bp fragment from the Bacillus subtilis genome.
yqiT EX: ccggaattctctagaatggaactttttaaatatatggaacgatacg(bold text is a sequence binding yqiT gene)
yqiT SP: ctgcagcggccgctactagtattagcgacgacttaaaatatgttgg(bold text is a sequence binding yqiT gene)
PCR protocol
The following was mixed in a PCR tube:
1ul 20uM yqiT EX primer
1ul 20uM yqiT SP primer
2ul 10X Pfu Ultra buffer
1.6ul dNTP mix
0.15ul Bacillus subtilis genome
0.5ul Pfu Ultra enzyme
13.75ul MilliQ
Performed PCR using the following program:
1. 95°C 2 minutes
2. 95°C 30 seconds
3. 50°C 30 seconds
4. 72°C 40 seconds
5. Repeat 2-4 29 times
6. 25°C forever
Purified PCR product using the Promega PCR purification kit.
Ran on gel to gel purify:
Lanes 6:marker 6ul
Lanes 2, 4:yqiT PCR product 10ul + 10xLoading Dye 2ul

PCR successful
Digestion
Digested purified PCR product and the lacI regulated promoter both with XbaI/PstI:
yqiT
3ul PCR product
1ul High buffer
0.5ul XbaI
0.5ul PstI
5ul MilliQ
lacI regulated promoter
4ul DNA
4ul High buffer
2ul XbaI
2ul PstI
28ul MilliQ
Incubated mixtures at 37°C for 1 hour.
Ran on gel to gel purify:
Lanes 3:marker 6ul
Lanes 2, 5:lacI regulated promoter 40ul + 10xLoading Dye 8ul
Lanes 1, 4:yqiT 10ul + 10xLoading Dye 2ul

Cut out the band and dissolved the gel using the Promega gel purification kit to extract DNA from.
Ligation
Ligated gel purified yqiT and vector with TaKaRa Solutions 1:
lacI regulated promoter (vector) 0.5ul
yqiT 5.5ul
TaKaRa Solutions 1 4ul
Incubated mixtures at 37°C for 10 minutes.
Transformation
7/5
TA cloning of YqiT
PCR of YqiT
0.25ul Takara ExTaq
5ul 10xExTaq Buffer
4ul dNTPmix
0.5ul Bacillus subtilis genome
0.5ul 5'primer(It is the same sequence as that on 6/21)
0.5ul 3'primer(It is the same sequence as that on 6/21)
up to 50ul MilliQ
Performed PCR using the following program:
1. 94°C 2 minutes
2. 94°C 30 seconds
3. 52°C 80 seconds
4. 72°C 60 seconds
5. Repeat 2-4 29 times
6. 25°C forever
lane1:marker
lane2~5:YqiT

PCR successful!
But we couldn't do ligation because TA cloning kit was not left.
We purified PCR products and put them in -20°C.
7/19
YqiT colony PCR
put a small amount of single colony into each tube with 5ul MilliQ water
↓
95°C 5min
↓
PCR reaction
1ul 10×buffer
0.8ul 2.5mMdNTP
0.08ul Ex-Taq
0.1ul 5’-primer
0.1ul 3’-primer
2.92ul MilliQ water
↓
added the PCR reaction to each tube.
↓
Performed PCR using the following program:
1. 95°C 2min
2. 95°C 30sec
3. 52°C 30sec
4. 72.5°C 80sec
5. repeat 2-4 29times
6. 25°C forever
PCR successful!
added a small amount of colony number 2 and 3 to each test tube with 4ml LB broth.
cultured them over night.
7/20
Miniprep of E.coli cells containing YqiT gene with Promega, Wizard Plus SV Miniprep DNA Purification System
7/27
sequencing YqiT by BIG DYE
1.8ul 5xB.D.3.1.buffer
0.4ul B.D.3.1.
6.3ul MilliQ water
1ul plasmid(0.15ug/ul)
0.5ul 5'or3'primer(3.2pmol/ul)
↓
PCR Program
1.96°C 2min
2.96°C 10sec
3.55°C 5sec
4.60°C 3min
5.go to 2.29times
6.25°C forever
↓
add 0.5ul PHOSPHATASE ALKALINE shrimp
↓
37°C 1hr incubate
↓
add 1ul 3MNaOAc
↓
add 25ul EtOH
↓
20000xg 4°C 10min centrifugation
↓
put off supernatant
↓
dry tubes
↓
add 15ul HiDi
↓
put them in the sequence machine
7/30~8/2
sequence YqiT by BigDye
unsuccessful
8/3
sequence YqiT by BigDye
successful
8/7~9
Digestion
insert YqiT gene into a terminator of iGEM parts
terminator
double terminator Miniprep., using Promega kit
plate1 23L
8/12~14
PCR of YqiT for restriction enzyme fragmentation and infusion
PCR protocol
The following was mixed in a PCR tube:
0.2ul 100uM yqiT EX primer/yqiT infusion 5'primer
0.2ul 100uM yqiT SP primer/yqiT infusion 3'primer
2ul 10X Pfu Ultra buffer
1.6ul dNTP mix
0.2ul plasmid containing YqiT gene(that sequence matches YqiT gene of Batillus genome)
0.5ul Pfu Ultra enzyme
15.3ul MilliQ
Performed PCR using the following program:
1. 95°C 2 minutes
2. 95°C 30 seconds
3. 55°C 30 seconds
4. 72.5°C 30 seconds
5. Repeat 2-4 29 times
6. 25°C forever
restriction enzyme fragmentation of double terminator by XbaI
1ug plasmid
10ul BSA
10ul 10xM buffer
2.5ul XbaI
up to 100ul MilliQ water
37°C over night
restriction enzyme fragmentation of double terminator by EcoRI
1ug plasmid
10ul 10xH buffer
2.5ul EcoRI
up to 100ul MilliQ water
37°C over night
restriction enzyme fragmentation of YqiT gene by SpeI
1ug DNA
10ul 10xH buffer
2.5ul SpeI
up to 100ul MilliQ water
37°C over night
restriction enzyme fragmentation of YqiT gene by EcoRI
1ug DNA
10ul 10xH buffer
2.5ul EcoRI
up to 100ul MilliQ water
37°C over night
8/15,16
ligate YqiT and double terminator
August
Aim
We decided to use YqiT as a reporter of dioxin!
So, we began to create this construct
1.-XRE-Gal1 promoter-yqiT-
2.-Ahr-←Gal1promoter/Gal10 promoter→-Arnt-
- PCR of yqiT and insert it in iGEM parts(plate 1-7D:Gal1 promoter)
Performed PCR using the following program:
1. 95°C 2 minutes
2. 95°C 30 seconds
3. 55°C 30 seconds
4. 72.5°C 30 seconds
5. Repeat 2-4 29 times
6. 25°C forever
- cut YqiT off with EcoRI and SpeI
- cut P1-7D off with EcoRI and XbaI
- ligate the upper two DNA
September
- read the sequence of Gal1 promoter+yqiT
→the result conforms with NCBI datebase
- insert XRE in P1-7D including yqiT using infusion kit(clonthech)
Aim
TA cloning of Ahr, Arnt and gal1/10 promoter
- PCR of Ahr and Arnt for TA cloning(Ex-taq , Funakoshi)
template:human Ahr cDNA and human Arnt cDNA
1,95°C 2min
2,95°C 30sec
3,55°C 30sec
4,72.5°C 1.5min
5,go°C to 2, 29times
6,25°C forever
==October='em
- PCR of gal1/10 promoter (Ex-taq , Funakoshi)
template:S.serevisiae genome DNA
1,95°C 2min
2,95°C 30sec
3,55°C 30sec
4,72.5°C1min
5,go°C to 2, 29times
6,25°C forever
- TA cloning of Ahr, Arnt and gal1/10 promoter
ligate PCR product and T bector
↓
put each plasmid into different E.coli
↓
Miniprep of them by using Promega kit
↓
read each sequence in order to check whether there is plasmids including Ahr, Arnt or gal1/10 promoter
Aim
- check YqiT expression
Using iGEM parts, create the construct that produces YqiT steadily or reguratory
P2-10F-YqiT-double terminator
P2-16E-YqiT-double terminator
P1-22A-YqiT-double terminator
P1-21B-YqiT-double terminator
cut YqiT-double terminator off with XbaI and PstI
cut P2-10F, P2-16E, P1-22A and P1-21B off with SpeI and PstI
↓
ligate YqiT-double terminator and iGEM part
↓
put each plasmid into different E.coli
↓
Miniprep of them by using Promega kit
↓
read each sequence in order to check whether there is plasmids including the construct
| Home | The Team | The Project | Parts Submitted to the Registry | Modeling | Notebook | Protocols | Ethics |
|---|