From 2009.igem.org
(Difference between revisions)
|
|
| Line 18: |
Line 18: |
| | val_func(); | | val_func(); |
| | ~val_func(); | | ~val_func(); |
| - | int type; //the type of the kineticlaw | + | int type; //the type of the kineticlaw |
| - | //Due to the ASTNode(a simple kind of data structure)
| + | //Due to the ASTNode(a simple kind of data structure) |
| - | //used to store mathematical expressions in SBML
| + | //used to store mathematical expressions in SBML |
| - | //most of them is algebraic expression), it is hard
| + | //most of them is algebraic expression), it is hard |
| - | //to judge the type mentioned in the file. Usually we
| + | //to judge the type mentioned in the file. Usually we |
| - | //give a number 15 to this variable, which implies the
| + | //give a number 15 to this variable, which implies the |
| - | //expression will be execute by route “user define”.
| + | //expression will be execute by route “user define”. |
| | | | |
| - | int spec_num; //number of species | + | int spec_num; //number of species |
| - | int para_num; //number of parameters | + | int para_num; //number of parameters |
| - | vector<int> spec; //serial number of the species of the reaction | + | vector<int> spec; //serial number of the species of the reaction |
| - | vector <double> para; //initial value of the parameters | + | vector <double> para; //initial value of the parameters |
| - | vector <string> para_name; //IDs/names of the parameters | + | vector <string> para_name; //IDs/names of the parameters |
| - | vector <string> var_name; //IDs/names of the species | + | vector <string> var_name; //IDs/names of the species |
| - | map<int,double> min_data; //N/A | + | map<int,double> min_data; //N/A |
| - | map<int , double> max_data; //N/A | + | map<int , double> max_data; //N/A |
| - | const ASTNode * head; //ASTNode tree of the formula | + | const ASTNode * head; //ASTNode tree of the formula |
| - | double value( const vector<double>& s); //N/A | + | double value( const vector<double>& s); //N/A |
| | }; | | }; |
| | </pre> | | </pre> |
Revision as of 14:03, 20 October 2009

SBML
[http://sbml.org The Systems Biology Markup Language (SBML)] is a computer-readable format for representing models of biological processes. It's applicable to simulations of metabolism, cell-signaling, and many other topics. SBML has been evolving since mid-2000 thanks to an international community of software developers and users. Thus, we make SBML to access our software, which makes it more compatible to other biological projects.
To access the requirement, there must be a tool helps us to fetch the list of data from a SBML file. Fortunately, [http://sbml.org/Software/libSBML LibSBML] assists us for all the SBML related staff. LibSBML is an open-source programming library to help you read, write, manipulate, translate, and validate SBML files and data streams. The Latest stable release of LibSBML is 4.0.0. In terms of our software, the following two major functions has been accomplished based on LibSBML.
1.ReadSBMLFile
The class ReadSBMLFile is used to extract all useful data from a SBML file and send them to the class “val_func”. Since each class represents one term of the the expression. all the terms will be push into a vector. In another word, another container has been created used to place a list of "val_func", of course you can realize that Nth list of "val_func" accommodates all the informations related to Nth species in this Biological system. Finally, all the containers will be package orderly and send to the Particle Swarm Optimization Algorithm (PSO) part or the Global Sensitivity Analysis (GSA) part for the next step. Here are the components of class “val_func”.
class val_func
{
public:
val_func();
~val_func();
int type; //the type of the kineticlaw
//Due to the ASTNode(a simple kind of data structure)
//used to store mathematical expressions in SBML
//most of them is algebraic expression), it is hard
//to judge the type mentioned in the file. Usually we
//give a number 15 to this variable, which implies the
//expression will be execute by route “user define”.
int spec_num; //number of species
int para_num; //number of parameters
vector<int> spec; //serial number of the species of the reaction
vector <double> para; //initial value of the parameters
vector <string> para_name; //IDs/names of the parameters
vector <string> var_name; //IDs/names of the species
map<int,double> min_data; //N/A
map<int , double> max_data; //N/A
const ASTNode * head; //ASTNode tree of the formula
double value( const vector<double>& s); //N/A
};
2.WriteSBMLFile
The class WriteSBMLFile is used to output a SBML file base on the species and parameters selected from the database part.
In this part, the class WriteSBMLFile extract data from the two following file: “subs.dat”; ”dbt.dat”.
Assitant Input Plug-ins
In order to simplify the input procedure of the data file describing time course which sometimes gets really redundant, we also program a small .exe file to help convert *.bmp file to *.dat file based on following assumptions.
- The x-axis and y-axis are already unified to the range [0,1], while any other range could be realized by multiple additional factors on the unified range.
- The program will only identify black pixels on the picture, and for any given x value, the y value is decided by the average of all existing points corresponding to that same x value.
- Color depth sould be no less than 8 bits.
- Sampling step length is determined by 1 divided by the horizontal pixel numbers.
Following is a small demo of this tool.
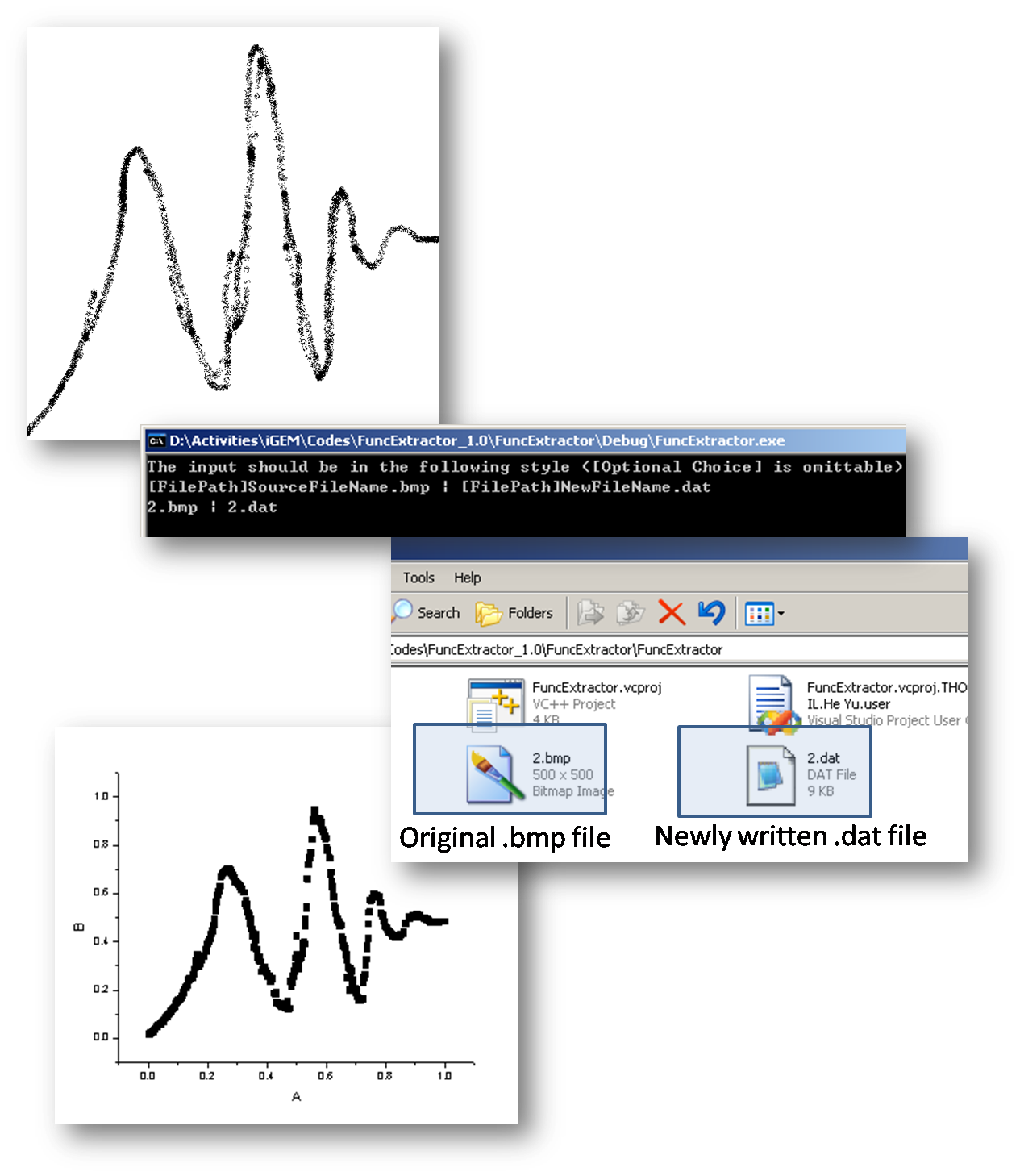
There are definitely quite a lot to improve, including
1.Rescale-able range of x-axis and y-axis
2.User designated step length
3.Function of ‘smoothing’
4.Identify the curve based on contrast rather than requiring the effective pixel to be black.
 "
"
