 "
"
Team:EPF-Lausanne/Theory
From 2009.igem.org
(Difference between revisions)
(→Molecular dynamics theory) |
(→Molecular dynamics theory) |
||
| Line 7: | Line 7: | ||
Molecular dynamics simulation consists of the numerical, step-by-step, solution of the classical equations of motion. For this purpose we need to be able to calculate the forces acting on the atoms, and these are usually derived from a potential energy. This potential energy can be divided into: | Molecular dynamics simulation consists of the numerical, step-by-step, solution of the classical equations of motion. For this purpose we need to be able to calculate the forces acting on the atoms, and these are usually derived from a potential energy. This potential energy can be divided into: | ||
* '''the non-bonded interactions''': | * '''the non-bonded interactions''': | ||
| - | **The ''Lennard-Jones potential'' is the most commonly used form, with two parameters: σ, the diameter, and ε, the well depth. It takes into account the Van der Waals forces. It represents the non-bonded forces and the total potential energy can be calculated from the sum of energy contributions between pairs of atoms. [[Image:lennard_jones_vdw_forces.jpg | + | **The ''Lennard-Jones potential'' is the most commonly used form, with two parameters: σ, the diameter, and ε, the well depth. It takes into account the Van der Waals forces. It represents the non-bonded forces and the total potential energy can be calculated from the sum of energy contributions between pairs of atoms. [[Image:lennard_jones_vdw_forces.jpg|center]] |
**when electrostatic charges are present, we add the ''Coulomb force'', where Q1, Q2 are the charges and ϵ0 is the permittivity of free space | **when electrostatic charges are present, we add the ''Coulomb force'', where Q1, Q2 are the charges and ϵ0 is the permittivity of free space | ||
[[Image:Coulomb.jpg|200px|center]] | [[Image:Coulomb.jpg|200px|center]] | ||
| Line 13: | Line 13: | ||
* '''the bonded interactions''': angles, bonds and dihedral angles have to be taken into account | * '''the bonded interactions''': angles, bonds and dihedral angles have to be taken into account | ||
| - | [[Image:bonded.jpg|400px | + | [[Image:bonded.jpg|400px|center]] |
Revision as of 11:08, 27 July 2009

Contents |
Molecular dynamics theory
Molecular dynamics simulation consists of the numerical, step-by-step, solution of the classical equations of motion. For this purpose we need to be able to calculate the forces acting on the atoms, and these are usually derived from a potential energy. This potential energy can be divided into:
- the non-bonded interactions:
- The Lennard-Jones potential is the most commonly used form, with two parameters: σ, the diameter, and ε, the well depth. It takes into account the Van der Waals forces. It represents the non-bonded forces and the total potential energy can be calculated from the sum of energy contributions between pairs of atoms.
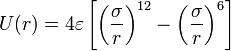
- when electrostatic charges are present, we add the Coulomb force, where Q1, Q2 are the charges and ϵ0 is the permittivity of free space
- The Lennard-Jones potential is the most commonly used form, with two parameters: σ, the diameter, and ε, the well depth. It takes into account the Van der Waals forces. It represents the non-bonded forces and the total potential energy can be calculated from the sum of energy contributions between pairs of atoms.


- the bonded interactions: angles, bonds and dihedral angles have to be taken into account

Too understand a bit more, you can see the following article:
Introduction to Molecular Dynamics Simulation - Michael P. Allen