 "
"
Team:EPF-Lausanne/Modeling/Simulation
From 2009.igem.org
m (→Creation of the pdb file) |
|||
| (26 intermediate revisions not shown) | |||
| Line 3: | Line 3: | ||
</div><div CLASS="epfl09"> | </div><div CLASS="epfl09"> | ||
| - | = | + | |
| + | |||
| + | <html> | ||
| + | <body> | ||
| + | <form action="input_button.htm"> | ||
| + | <p align="right"> | ||
| + | <input type="button" name="lien" value="Click here to return to Analysis methods" | ||
| + | onClick="self.location.href='https://2009.igem.org/Team:EPF-Lausanne/Analysis_methods'"> | ||
| + | </p> | ||
| + | </form> | ||
| + | </body> | ||
| + | </html> | ||
| + | |||
| + | = Creating input files for namd = | ||
| Line 9: | Line 22: | ||
We open VMD, and lauch our protein. | We open VMD, and lauch our protein. | ||
| + | |||
| + | |||
| + | ==Nomenclature== | ||
| + | Here are the convention we are using to name files: | ||
| + | :PROT.pdb : pdb of the protein downloaded from the internet(in capital letters) | ||
| + | :prot.pdb : pdb after removing unwanted atoms (step 3 on this page) | ||
| + | :protp.pdb : pdb after processing by psfgen | ||
| + | :protp.psf : psf after processing by psfgen | ||
| + | :protp_wb.pdb : pdb after solvating | ||
| + | :protp_wb.psf : psf after solvating | ||
| + | :protp_wb_i.pdb : pdb after adding ions (final version of the file) | ||
| + | :protp_wb_i.psf : psf after adding ions (final version of the file) | ||
| + | |||
| Line 24: | Line 50: | ||
We have created the file 2v0u.pdb, which contains the coordinates of the protein alone without hydrogens. | We have created the file 2v0u.pdb, which contains the coordinates of the protein alone without hydrogens. | ||
Quit VMD. | Quit VMD. | ||
| + | |||
| + | |||
==Creation of the psf file== | ==Creation of the psf file== | ||
::''.pdb file + topology file (*.rtf) → .psf file + matching .pdb file'' | ::''.pdb file + topology file (*.rtf) → .psf file + matching .pdb file'' | ||
| - | |||
::.psf file contains atom parameters derived from topology, but is not human readable. | ::.psf file contains atom parameters derived from topology, but is not human readable. | ||
| - | |||
There are 2 ways to create a psf: | There are 2 ways to create a psf: | ||
| - | * in VMD, Extensions → Modeling → Automatic PSF Builder (WARNING: bug prevent from dealing with multile | + | * in VMD, Extensions → Modeling → Automatic PSF Builder (psfgen GUI) (WARNING: bug prevent from dealing with multile chains!) |
| + | * We first make a pgn file, which is a script file that will command psfgen. | ||
| - | + | ===Unique segment:=== | |
| - | In a Terminal window, open a text editor and create a file that you'll call | + | In a Terminal window, open a text editor and create a file that you'll call 2v0w.pgn: |
: package require psfgen | : package require psfgen | ||
: topology [[Media:Top_all27_prot_lipid-fmc.txt|top_all27_prot_lipid.inp]] | : topology [[Media:Top_all27_prot_lipid-fmc.txt|top_all27_prot_lipid.inp]] | ||
| + | |||
: pdbalias residue HIS HSE | : pdbalias residue HIS HSE | ||
: pdbalias atom ILE CD1 CD | : pdbalias atom ILE CD1 CD | ||
| + | |||
: segment U | : segment U | ||
| - | : {pdb | + | : {pdb 2v0w.pdb} |
| - | : coordpdb | + | : coordpdb 2v0w.pdb U |
| + | |||
: guesscoord | : guesscoord | ||
| - | : writepdb | + | : writepdb 2v0wp.pdb |
| - | : writepsf | + | : writepsf 2v0wp.psf |
| - | You'll need to place the topology file within same directory. Here is the current [[Media:Top_all27_prot_lipid-fmc.txt|topology file]], please rename to Top_all27_prot_lipid-fmc.rtf after download. | + | You'll need to place the topology file within same directory. Here is the current [[Media:Top_all27_prot_lipid-fmc.txt|topology file]], please rename to Top_all27_prot_lipid-fmc.rtf after download. The topology file is specific for the .pdb we have, ask if you need one for dark/light FMN. |
In a Terminal window, type the following command: | In a Terminal window, type the following command: | ||
| - | : > vmd -dispdev text -e | + | : > vmd -dispdev text -e 2v0w.pgn |
| - | This will run the | + | This will run the psfgen from the file 2v0w.pgn and generate the psf (2v0wp.psf) and the pdb (2v0wp.pdb) file of the protein with hydrogens. |
A new pdb file with the complete coordinates of all atoms is written, including H; and a psf file with the complete structural information of the protein. | A new pdb file with the complete coordinates of all atoms is written, including H; and a psf file with the complete structural information of the protein. | ||
| + | |||
===Case of multiple segments:=== | ===Case of multiple segments:=== | ||
| Line 58: | Line 89: | ||
This section is based on [[Media:Ug.pdf|psfgen manual]]. | This section is based on [[Media:Ug.pdf|psfgen manual]]. | ||
| - | Psfgen is not able to deal with multiple sequences within a single .pdb (even using TER and different seg | + | Psfgen is not able to deal with multiple sequences within a single .pdb (even using TER and different seg names). We have to separate them, either by hand, either using grep. So you should have as many .pdb files as chains (2v0u_prot.pdb, 2v0u_fmn.pdb). |
| - | and .pgn gets | + | and .pgn gets. I added some comments to explain the commands(//), please remove them before run. |
| - | :package require psfgen | + | :package require psfgen //check if plugin available |
| - | :topology [[Media:Top_all27_prot_lipid-fmn_dark.txt| | + | :topology [[Media:Top_all27_prot_lipid-fmn_dark.txt|top_all27_prot_lipid-fmn_dark.inp]] //loads topology file |
| - | :pdbalias residue HIS HSE | + | :pdbalias residue HIS HSE //some aliases |
| - | :pdbalias atom ILE CD1 CD | + | :pdbalias atom ILE CD1 CD //some aliases |
| - | :segment 2v0u { | + | :segment 2v0u { //creates a chain that'll contain the prot |
| - | :pdb 2v0u_prot.pdb | + | :pdb 2v0u_prot.pdb //loads list of atoms |
:} | :} | ||
| - | :coordpdb 2v0u_prot.pdb 2v0u | + | :coordpdb 2v0u_prot.pdb 2v0u //loads coord. of atoms |
| - | :segment fmn { | + | :segment fmn { //creates the second chain |
| - | :pdb 2v0u_fmn.pdb | + | :pdb 2v0u_fmn.pdb //loads atoms from FMN |
| - | :first none | + | :first none //prevents psfgen from "capping" the first residue |
| - | :last none | + | :last none //prevents psfgen from "capping" the last residue |
:} | :} | ||
| - | :coordpdb 2v0u_fmn.pdb fmn | + | :coordpdb 2v0u_fmn.pdb fmn //loads coord. of atoms |
| - | :guesscoord | + | :guesscoord //complete missing coord. |
| - | :writepdb 2v0up.pdb | + | :writepdb 2v0up.pdb //write .pdb containing all previously loaded chains |
| - | :writepsf 2v0up.psf | + | :writepsf 2v0up.psf //write .psf containing all previously loaded chains |
| - | == | + | The structure is quite important, as psfgen applies some patch at the end of loaded chains (NTERM, CTERM). In the case of FMN without link to the cystein, we have to prevent psfgen from adding atoms to neutralize the molecule as if it was a protein. That's the purpose of ''first none'' and ''last none''. |
| - | ::''.pdb containing the protein → .pdb containing the protein and water molecules'' | + | |
| + | |||
| + | |||
| + | ==Solvating the protein == | ||
| + | ::''.pdb + .psf containing the protein → .pdb + .psf containing the protein and water molecules'' | ||
| Line 92: | Line 127: | ||
In the VMD Main window, open the Tk Console, and type: | In the VMD Main window, open the Tk Console, and type: | ||
: package require solvate | : package require solvate | ||
| - | : solvate | + | : solvate 2v0up.psf 2v0up.pdb -t 12 -o 2v0up_wb |
| - | + | ||
| - | + | ||
| + | The "solvate package" will put the protein in a box of water. The -t option creates the water box dimensions such that there is a layer of water 12 Angström in each direction from the atom with the largest coordinate in that direction. We have to take a special care to '''be sure we don't have interaction''' with the protein in the next periodic box. Minimization usually shrinks the water box. Minimal distance between nearer atoms of 2 boxes should be higher than ~12 Angstroem. As we have a small protein, we can increase the size of the box without having too many atoms. The -o option creates the output files our_prot_in_a_water_box .pdb and our_prot_in_a_water_box.psf. | ||
| - | == | + | |
| - | ::''add ions in .pdb'' | + | |
| + | ==Add ions == | ||
| + | ::''add ions in .pdb + .psf'' | ||
In VMD, we load the psf and the pdb created with the pgn. Under Extension/Modeling/Add Ions, and knowing the charge of the protein (for exemple -7), we add 7 atoms of Na. | In VMD, we load the psf and the pdb created with the pgn. Under Extension/Modeling/Add Ions, and knowing the charge of the protein (for exemple -7), we add 7 atoms of Na. | ||
Instead of concentrations, we click user defined to add 7 Na, and neutralize. | Instead of concentrations, we click user defined to add 7 Na, and neutralize. | ||
| + | The automatic mode works quite well. At least it is really efficient to calculate the total charge of the system, but it might add both positive and negative charges to reach neutral potential. So we prefer to add a given number of ions by hand. | ||
[[Image:Ionization.jpg|frame|center|Ionization]] | [[Image:Ionization.jpg|frame|center|Ionization]] | ||
| - | == | + | |
| + | ==Measurement of the water box coordinates== | ||
::''we need the origin and 3 dimension vectors of the system as initial conditions for namd'' | ::''we need the origin and 3 dimension vectors of the system as initial conditions for namd'' | ||
| - | In | + | In VMD, load 2v0up_wb_i.psf and 2v0up_wb_i.pdb. This will display our protein in a water box. |
In the VMD TkCon window type: | In the VMD TkCon window type: | ||
: set everyone [atomselect top all] | : set everyone [atomselect top all] | ||
: measure minmax $everyone | : measure minmax $everyone | ||
| + | |||
This gives the minimum and maximum values of x, y and z coordinates of the entire protein-water system, relative to the origin of the coordinate system. | This gives the minimum and maximum values of x, y and z coordinates of the entire protein-water system, relative to the origin of the coordinate system. | ||
| Line 120: | Line 159: | ||
: measure center $everyone | : measure center $everyone | ||
| - | These coordinates have to be kept and recorded for the referential. | + | These coordinates have to be kept and recorded for the referential of namd. |
| + | |||
| + | You now have all the input files for namd! | ||
| - | |||
| - | == | + | = More informations from the tutorial = |
| + | ==NOT NEEDED! Simulation with Periodic Boundary Conditions == | ||
::''.psf + .pdb + .prm + .conf → namd → anything you want'' | ::''.psf + .pdb + .prm + .conf → namd → anything you want'' | ||
| + | This part of the tutorial doesn't have its place here anymore, as we have to detail much more the process of launching namd. Please go back to the [[Team:EPF-Lausanne/Implementation|root]] for more informations. | ||
The use of periodic boundary conditions are effective in eliminating surface interaction of the water molecules and creating a more faithful representation of the in vivo environment than a water sphere surrounded by vacuum provides. | The use of periodic boundary conditions are effective in eliminating surface interaction of the water molecules and creating a more faithful representation of the in vivo environment than a water sphere surrounded by vacuum provides. | ||
| Line 141: | Line 183: | ||
| - | == | + | |
| - | Open VMD, load first the file | + | ==VMD - Make a movie from the .dcd== |
| + | This step occurs after the namd calculation. | ||
| + | Open VMD, load first the file 2v0up_wb_i.psf, and then the file .dcd automatically created. The protein will appear surrounded by molecules of water. | ||
To see it more clearly, go in Graphics → Representation, and create 3 replicates. | To see it more clearly, go in Graphics → Representation, and create 3 replicates. | ||
| Line 156: | Line 200: | ||
[[Image: Movie_generation.jpg|frame|center]] | [[Image: Movie_generation.jpg|frame|center]] | ||
| - | |||
| - | |||
| - | |||
| - | |||
Latest revision as of 15:23, 19 October 2009

Creating input files for namd
We start with the PDB file of the protein, obtained through the Protein Data Bank.
We open VMD, and lauch our protein.
Nomenclature
Here are the convention we are using to name files:
- PROT.pdb : pdb of the protein downloaded from the internet(in capital letters)
- prot.pdb : pdb after removing unwanted atoms (step 3 on this page)
- protp.pdb : pdb after processing by psfgen
- protp.psf : psf after processing by psfgen
- protp_wb.pdb : pdb after solvating
- protp_wb.psf : psf after solvating
- protp_wb_i.pdb : pdb after adding ions (final version of the file)
- protp_wb_i.psf : psf after adding ions (final version of the file)
Creation of the pdb file
- .pdb from the web → .pdb file ready for vmd processing (removing some atoms)
After creating the molecule containing the .pdb downloaded from internet, in the Tk Console menu of VMD we open the VMD TkCon window, and type the following commands:
- set our_protein [atomselect top "not water and not resname GOL"]
- $our_protein writepdb 2v0u.pdb
This will select all the protein except the water and except the glycerol, with the cofactor. If we have multiple sequences, we can simply select them and write them to different .pdb.
We have created the file 2v0u.pdb, which contains the coordinates of the protein alone without hydrogens. Quit VMD.
Creation of the psf file
- .pdb file + topology file (*.rtf) → .psf file + matching .pdb file
- .psf file contains atom parameters derived from topology, but is not human readable.
There are 2 ways to create a psf:
- in VMD, Extensions → Modeling → Automatic PSF Builder (psfgen GUI) (WARNING: bug prevent from dealing with multile chains!)
- We first make a pgn file, which is a script file that will command psfgen.
Unique segment:
In a Terminal window, open a text editor and create a file that you'll call 2v0w.pgn:
- package require psfgen
- topology top_all27_prot_lipid.inp
- pdbalias residue HIS HSE
- pdbalias atom ILE CD1 CD
- segment U
- {pdb 2v0w.pdb}
- coordpdb 2v0w.pdb U
- guesscoord
- writepdb 2v0wp.pdb
- writepsf 2v0wp.psf
You'll need to place the topology file within same directory. Here is the current topology file, please rename to Top_all27_prot_lipid-fmc.rtf after download. The topology file is specific for the .pdb we have, ask if you need one for dark/light FMN.
In a Terminal window, type the following command:
- > vmd -dispdev text -e 2v0w.pgn
This will run the psfgen from the file 2v0w.pgn and generate the psf (2v0wp.psf) and the pdb (2v0wp.pdb) file of the protein with hydrogens. A new pdb file with the complete coordinates of all atoms is written, including H; and a psf file with the complete structural information of the protein.
Case of multiple segments:
This section is based on psfgen manual.
Psfgen is not able to deal with multiple sequences within a single .pdb (even using TER and different seg names). We have to separate them, either by hand, either using grep. So you should have as many .pdb files as chains (2v0u_prot.pdb, 2v0u_fmn.pdb).
and .pgn gets. I added some comments to explain the commands(//), please remove them before run.
- package require psfgen //check if plugin available
- topology top_all27_prot_lipid-fmn_dark.inp //loads topology file
- pdbalias residue HIS HSE //some aliases
- pdbalias atom ILE CD1 CD //some aliases
- segment 2v0u { //creates a chain that'll contain the prot
- pdb 2v0u_prot.pdb //loads list of atoms
- }
- coordpdb 2v0u_prot.pdb 2v0u //loads coord. of atoms
- segment fmn { //creates the second chain
- pdb 2v0u_fmn.pdb //loads atoms from FMN
- first none //prevents psfgen from "capping" the first residue
- last none //prevents psfgen from "capping" the last residue
- }
- coordpdb 2v0u_fmn.pdb fmn //loads coord. of atoms
- guesscoord //complete missing coord.
- writepdb 2v0up.pdb //write .pdb containing all previously loaded chains
- writepsf 2v0up.psf //write .psf containing all previously loaded chains
The structure is quite important, as psfgen applies some patch at the end of loaded chains (NTERM, CTERM). In the case of FMN without link to the cystein, we have to prevent psfgen from adding atoms to neutralize the molecule as if it was a protein. That's the purpose of first none and last none.
Solvating the protein
- .pdb + .psf containing the protein → .pdb + .psf containing the protein and water molecules
Now, the protein needs to be solvated, i.e., put inside water, to more closely resemble the cellular environment. This will be done by placing the protein in a water box, in preparation for minimization and equilibration with periodic boundary conditions.
In the VMD Main window, open the Tk Console, and type:
- package require solvate
- solvate 2v0up.psf 2v0up.pdb -t 12 -o 2v0up_wb
The "solvate package" will put the protein in a box of water. The -t option creates the water box dimensions such that there is a layer of water 12 Angström in each direction from the atom with the largest coordinate in that direction. We have to take a special care to be sure we don't have interaction with the protein in the next periodic box. Minimization usually shrinks the water box. Minimal distance between nearer atoms of 2 boxes should be higher than ~12 Angstroem. As we have a small protein, we can increase the size of the box without having too many atoms. The -o option creates the output files our_prot_in_a_water_box .pdb and our_prot_in_a_water_box.psf.
Add ions
- add ions in .pdb + .psf
In VMD, we load the psf and the pdb created with the pgn. Under Extension/Modeling/Add Ions, and knowing the charge of the protein (for exemple -7), we add 7 atoms of Na.
Instead of concentrations, we click user defined to add 7 Na, and neutralize.
The automatic mode works quite well. At least it is really efficient to calculate the total charge of the system, but it might add both positive and negative charges to reach neutral potential. So we prefer to add a given number of ions by hand.

Measurement of the water box coordinates
- we need the origin and 3 dimension vectors of the system as initial conditions for namd
In VMD, load 2v0up_wb_i.psf and 2v0up_wb_i.pdb. This will display our protein in a water box.
In the VMD TkCon window type:
- set everyone [atomselect top all]
- measure minmax $everyone
This gives the minimum and maximum values of x, y and z coordinates of the entire protein-water system, relative to the origin of the coordinate system.
The center of the water box may be determined by typing:
- measure center $everyone
These coordinates have to be kept and recorded for the referential of namd.
You now have all the input files for namd!
More informations from the tutorial
NOT NEEDED! Simulation with Periodic Boundary Conditions
- .psf + .pdb + .prm + .conf → namd → anything you want
This part of the tutorial doesn't have its place here anymore, as we have to detail much more the process of launching namd. Please go back to the root for more informations.
The use of periodic boundary conditions are effective in eliminating surface interaction of the water molecules and creating a more faithful representation of the in vivo environment than a water sphere surrounded by vacuum provides.
We first create a configuration file:
From the tutorial of VMD, we can download the configuration files for the minimization and equilibration of the protein in a water box. The process of editing .conf will be explained on a separate page.
Look at the tutorial for a more detailed explanation of the different parameters listed.
The simulation can be run by typing in a Terminal window:
- namd2 our_prot_configuration_file.conf > output_file.log
Output of the water sphere minimization-equilibration simulation will yield eleven output files. See the tutorial for a more detailed explanation of each file.
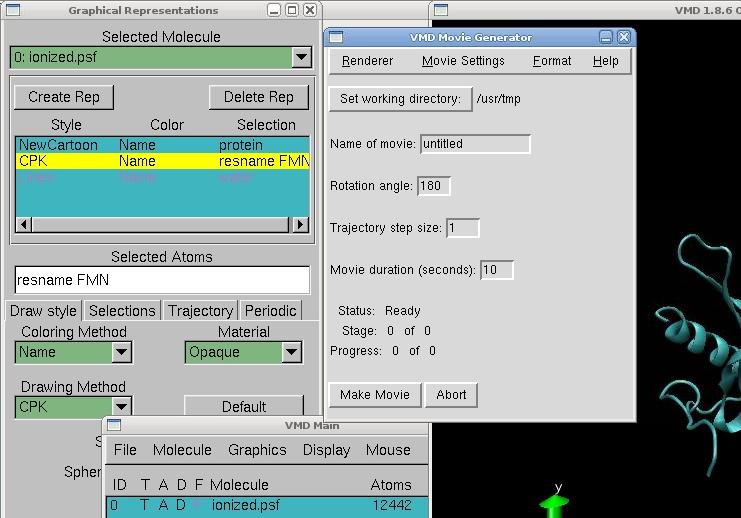
VMD - Make a movie from the .dcd
This step occurs after the namd calculation. Open VMD, load first the file 2v0up_wb_i.psf, and then the file .dcd automatically created. The protein will appear surrounded by molecules of water. To see it more clearly, go in Graphics → Representation, and create 3 replicates.
In the first replicates, type in selected atoms "protein" to select all residues in the protein, and choose NewCartoon as the drawing method for example. It will identify how many helices, betasheets and coils are present in the protein. In the second replicate, type "resname FMN" to select only the cofactor, and choose CPK for example to display the atoms of the cofactor. Finally, type "water" in the last replicate, and you can choose to deselect all molecules of water for a clearest view by double-clicking on it.
Now, to make a movie, just go on the VMD Main menu, and click on the triangle in the right bottom of the menu, and go to the Extension → Vizualization → Movie Maker Here, just change the name of the movie, the working directory and make the movie!

![]()