 "
"
Team:EPF-Lausanne/Modeling/Simulation
From 2009.igem.org
(→Simulation - Minimization) |
(→1. Creation of the pdb file) |
||
| Line 16: | Line 16: | ||
In the Tk Console menu of VMD we open the VMD TkCon window, and type the following commands: | In the Tk Console menu of VMD we open the VMD TkCon window, and type the following commands: | ||
| - | : set our_protein [atomselect top not water and not GOL] | + | : set our_protein [atomselect top "not water and not resname GOL"] |
: $our_protein writepdb our_protein.pdb | : $our_protein writepdb our_protein.pdb | ||
| Line 22: | Line 22: | ||
We have created the file our_protein.pdb, which contains the coordinates of the protein alone without hydrogens. | We have created the file our_protein.pdb, which contains the coordinates of the protein alone without hydrogens. | ||
Quit VMD. | Quit VMD. | ||
| - | |||
==2. Creation of the psf file== | ==2. Creation of the psf file== | ||
Revision as of 07:22, 27 July 2009

Contents |
Simulation - Minimization
We start with the PDB file of the protein, obtained through the Protein Data Bank.
We open VMD, and lauch our protein.
1. Creation of the pdb file
- .pdb from the web → .pdb file ready for vmd processing (removing some atoms)
In the Tk Console menu of VMD we open the VMD TkCon window, and type the following commands:
- set our_protein [atomselect top "not water and not resname GOL"]
- $our_protein writepdb our_protein.pdb
This will select all the protein except the water and except the glycerol, with the cofactor. We have created the file our_protein.pdb, which contains the coordinates of the protein alone without hydrogens. Quit VMD.
2. Creation of the psf file
- .pdb file + topology file (*.rtf) → .psf file + matching .pdb file
- .psf file contains atom parameters derived from topology, but is not human readable.
There are 2 ways to create a psf:
- in VMD, Extensions → Modeling → Automatic PSF Builder
- We first make a pgn file, which will be the target of psfgen.
In a Terminal window, open a text editor and type:
- package require psfgen
- topology top_all27_prot_lipid.inp
- pdbalias residue HIS HSE
- pdbalias atom ILE CD1 CD
- segment U
- {pdb our_protein.pdb}
- coordpdb our_protein.pdb U
- guesscoord
- writepdb our_protein_nv_pdb.pdb
- writepsf our_protein_nv_psf.psf
In a Terminal window, type the following command:
- > vmd -dispdev text -e our_protein.pgn
This will run the package psfgen on the file our_protein.pgn and generate the psf and the pdb file of the protein with hydrogens. A new pdb file with the complete coordinates of all atoms is written, including H; and a psf file with the complete structural information of the protein.
3. Solvating the Protein
- .pdb containing the protein → .pdb containing the protein and water molecules
Now, the protein needs to be solvated, i.e., put inside water, to more closely resemble the cellular environment. This will be done by placing the protein in a water box, in preparation for minimization and equilibration with periodic boundary conditions.
In the VMD Main window, open the Tk Console, and type:
- package require solvate
- solvate our_protein_nv_psf.psf our_protein_nv_pdb.pdb -t 5 -o our_prot_in_a_water_box
The "solvate package" will put the protein in a box of water. The -t option creates the water box dimensions such that there is a layer of water 5 Angström in each direction from the atom with the largest coordinate in that direction. The -o option creates the output files our_prot_in_a_water_box .pdb and our_prot_in_a_water_box.psf.
4. Add ions
- add ions in .pdb
In VMD, we load the psf and the pdb created with the pgn. Under Extension/Modeling/Add Ions, and knowing the charge of the protein (for exemple -7), we add 7 atoms of Na.
Instead of concentrations, we click user defined to add 7 Na, and neutralize.

5. Measurement of the water box coordinates
- we need the origin and 3 dimension vectors of the system as initial conditions for namd
In VMDload the our_prot_in_a_water_box.psf and the our_prot_in_a_water_box.pdb. This will display our protein in a water box.
In the VMD TkCon window type:
- set everyone [atomselect top all]
- measure minmax $everyone
This gives the minimum and maximum values of x, y and z coordinates of the entire protein-water system, relative to the origin of the coordinate system.
The center of the water box may be determined by typing:
- measure center $everyone
These coordinates have to be kept and recorded for the referential.
The necessary pdb and psf files must now be copied into a new folder, called common. We will store the files there so we have a single directory from which to access them, and so that we do not need to keep multiple copies of them.
6. Simulation with Periodic Boundary Conditions
- .psf + .pdb + .prm + .conf → namd → anything you want
The use of periodic boundary conditions are effective in eliminating surface interaction of the water molecules and creating a more faithful representation of the in vivo environment than a water sphere surrounded by vacuum provides.
We first create a configuration file:
From the tutorial of VMD, we can download the configuration files for the minimization and equilibration of the protein in a water box. The process of editing .conf will be explained on a separate page.
Look at the tutorial for a more detailed explanation of the different parameters listed.
The simulation can be run by typing in a Terminal window:
- namd2 our_prot_configuration_file.conf > output_file.log
Output of the water sphere minimization-equilibration simulation will yield eleven output files. See the tutorial for a more detailed explanation of each file.
7. Make a movie
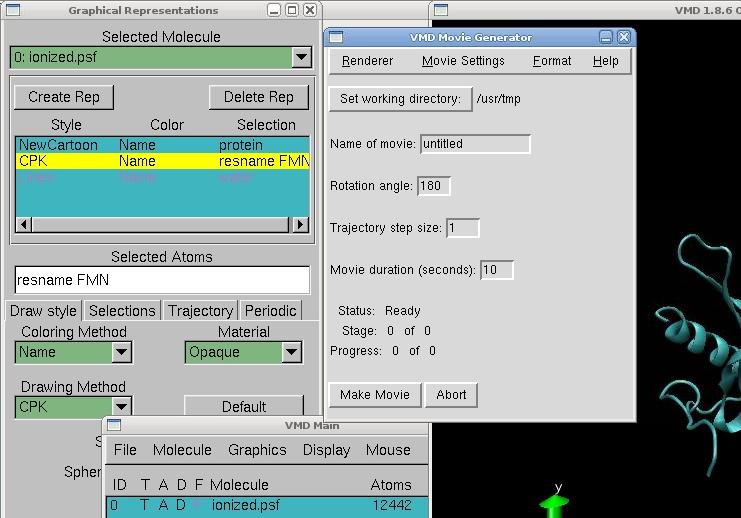
Open VMD, load first the file our_protein_ionized.psf, and then the file .dcd automatically created. The protein will appear surrounded by molecules of water. To see it more clearly, go in Graphics → Representation, and create 3 replicates.
In the first replicates, type in selected atoms "protein" to select all residues in the protein, and choose NewCartoon as the drawing method for example. It will identify how many helices, betasheets and coils are present in the protein. In the second replicate, type "resname FMN" to select only the cofactor, and choose CPK for example to display the atoms of the cofactor. Finally, type "water" in the last replicate, and you can choose to deselect all molecules of water for a clearest view by double-clicking on it.
Now, to make a movie, just go on the VMD Main menu, and click on the triangle in the right bottom of the menu, and go to the Extension → Vizualization → Movie Maker Here, just change the name of the movie, the working directory and make the movie!

Simulation - Equilibration
???