Procedures
Contents
- Polymerase Chain Reaction (PCR)
- Transformation
- Mini-Prep
- BioBrick Assembly
- Digestion
- Ligation
- Gel Electrophoresis
- PCB Extraction
- Glycerol Stock Solution
- Designing Primers
- Colony PCR
- Genomic DNA Purification
- Gel Extraction
- Making Competent Cells
- Tissue Flask Experiment
- R. shpaeroides Protocols and Recipes
Polymerase Chain Reaction (PCR)
- Polymerase Chain Reaction is used to amplify a small amount of purified DNA by several orders of magnitude. The key to PCR is thermal cycling which consists of three major steps which are repeated a number of times. The first is Denaturation which heats the DNA and splits it into two single stranded pieces. Next is the Annealing step where primers bind to each single stranded fragment. Finally Elongation takes place and Taq polymerase builds the entire strand of DNA from the primers. The process is exponential since each single strand of DNA becomes its own double strand creating a doubling effect. Only a few rounds of PCR are necessary to create large amounts of stock DNA. Note: This procedure assumes that you are starting with purified DNA.
Materials
- 5 uL AccuTaq LA 10X Buffer
- 2.5 uL dNTP mix
- x uL Template DNA (about 40ng/uL)
- 1 uL DMSO
- 1 uL Forward primer
- 1 uL Reverse Primer
- 0.5 uL AccuTaq LA DNA Polymerase
- dI H20 to 50 uL
Procedure
- Begin by combining the top 8 ingredients in a microcentrifuge tube-add water until the volume reaches 50 uL.
- Mix gently and briefly centrifuge to collect all components to the bottom of the tube. Add 50 uL of mineral oil to prevent evaporation.
- Before thermocycling beings start with one period of 30 seconds at 98C
- The following 3 step thermocycle should be repeated up to 30 times: 1) Denaturation 94C for 15 seconds 2) Annealing 65C for 20 seconds 3) Extension (Elongation) 68C for 20 minutes.
- Finish cycling with 68C for 10 minutes.
Back To Top
Transformation
- Transformation is the genetic alteration of a cell synthetically. In this case, plasmids are used to carry desired genes and coding sequences which are then taken up by the bacterial cells. Once in the cells, the plasmids are transcribed and translated into proteins in the same way as the host's genomic DNA. Transformations allow us to easily express things like antibiotic resistance and green florescent protein (GFP) in desired cells.
Materials
- dI H20
- Competent cells
- Plasmid DNA (to be taken up by cells)
- SOC media
- LB agar
- Antibiotics (vary depending on DNA used and are in LB plates)
Procedures
- Start thawing the competent cells on crushed ice.
- Add 50 µL of thawed competent cells and then 1 - 2 µL of the resuspended DNA to the labelled tubes. Make sure to keep the competent cells on ice.
- Incubate the cells on ice for 30 minutes.
- Heat shock the cells by immersion in a pre-heated water bath at 42ºC for 60 seconds. A water bath improves heat transfer to the cells.
- Incubate the cells on ice for 5 minutes.
- Add 200 μl of SOC broth (make sure that the broth does not contain antibiotics and is not contaminated)
- Incubate the cells at 37ºC for 2 hours while the tubes are rotating or shaking. Important: 2 hour recovery time helps in transformation efficiency, especially for plasmids with antibiotic resistance other than ampicillin.
- Label two petri dishes with LB agar and the appropriate antibiotic(s) with the part number, plasmid, and antibiotic resistance. Plate 20 µl and 200 µl of the transformation onto the dishes, and spread. This helps ensure that you will be able to pick out a single colony.
- Incubate the plate at 37ºC for 12-14 hours, making sure the agar side of the plate is up. If incubated for too long the antibiotics start to break down and un-transformed cells will begin to grow. This is especially true for ampicillin because the resistance enzyme is excreted by the bacteria, and inactivate the antibiotic outside of the bacteria.
- Note: Restriction sites E=EcoR1-HF; X=Xba1; S=Spe1; P=Pst1; M=Mixed Site
- To view the full BioBrick Manual procedures, please click [http://partsregistry.org/Help:Transformation_Protocol here].
Back To Top
Miniprep
- DNA Miniprep procedure is used to extract and isolate plasmid DNA from whole bacterial cells. This Miniprep kit was obtained from Sigma-Aldrich and exact solutions and formulas are unavailable (e.g. Wash Solution 1). To get exact ingredients please contact Sigma-Aldrich.
Materials
- Wash Solution 1
- Wash Solution 2
- dI H20
- Column Prep Solution
- Lyse Solution
- Elute solution
- Resuspension Solution
- RNase A solution
- Lysis Buffer
- Neutralization/Binding Buffer
- GenElute HP Binding Columns
Procedures
- Harvest Cells: Pellet 1-5mL of E. coli by centrifugation at 12,000RPM for 1 minute and discard supernatant.
- Resuspend Cells: Resuspend cells with 200uL of the Resuspension Solution containing RNase A solution.
- Cell Lysis: Lyse the resuspended cells by adding 200uL of Lysis buffer. Immediately mix by inverting the tube 6-8 times (Do Not Vortex). Do Not Allow Lysis to last longer than 5 minutes.
- Neutralization: Precipitate the cell debris by adding 350uL of the Neutralization/Binding buffer and mix by inverting the tube several times. Centrifuge at 12,000RPM for ten minutes. Cell debris, proteins, lipids, SDS and chromosomal DNA should fall out of solution.
- Prepare Column: Insert a GenElute Miniprep Binding Column into a microcentrifuge tube. Add 500uL of Column Preparation solution to each column and centrifuge at 12,000RPM for one minute and discard flow through liquid.
- Load Cleared Lysate: Transfer the cleared lysate from step 4 to the column and centrifuge at 12,000RPM for 1 minute and discard flow through liquid.
- Wash Column with Wash Solution 1: Add 500uL of Wash Solution 1 to the column and centrifuge at 12,000RPM for 1 minute and discard the flow through liquid.
- Wash Column with Wash Solution 2: Add 750uL of Wash Solution 2 to the column and centrifuge at 12,000RPM for 1 minute and discard the flow through liquid
- Centrifuge at 12,000RPM for 1 minute to remove excess ethanol.
- Elute DNA: Transfer column to fresh collection tube. Add 50uL of Elution Solution to the column. centrifuge at 12,000RPM for 1 minute. DNA is now present in the eluate and ready for immediate use or storage at -20C.
Back To Top
BioBrick Assembly
- BioBrick Assembly is a standard protocol for combining two BioBrick parts and positioning them in a destination plasmid. Specific cut sites for standard restriction enzymes are utilized to simplify the protocol and include EcoR1-HF, Xba1, Spe1 and Pst1. The assembly consists of two major parts: digestion and ligation. Note that BioBrick assembly requires purified, isolated DNA for all parts involved (upstream, downstream and destination plasmid). To view the full Biobrick Assembly manual, please click [http://ginkgobioworks.com/support/ here].
Materials
- NEBuffer 2
- BSA
- Restriction Enzymes (EcoR1-HF, Xba1, Spe1, Pst1)
- dI H20
- 10X T4 DNA Ligase Reaction Buffer
- 10X T4 DNA Ligase
- purified upstream BioBrick part
- purified downstream BioBrick part
- purified destination plasmid
Procedures
- Begin by thawing all DNA along with NEBuffer 2 and BSA.
- Label three separate PCR tubes upstream, downstream, and destination plasmid. To each tube 500ng of respective DNA and dilute to 42.5uL. Add 5uL of NEBuffer 2 and 0.5uL of BSA to each tube.
- Add 1uL of the first appropriate enzyme to each tube (see [http://ginkgobioworks.com/support/BioBrick_Assembly_Manual.jpg chart] for correct enzyme). Then add 1uL of second appropriate enzyme to each tube.
- Flick the tubes to mix contents and microcentrifuge for a few seconds to collect liquid in bottom of tube.
- Incubate the three digests at 80C for 20 minutes to deactivate the restriction enzymes. The digestion portion of the procedure is now completed. You may wish to store the products at -20C or proceed directly to the ligation step.
- Begin ligation by thawing 10X T4 DNA Ligase Reaction Buffer and enzyme. Agitate the buffer until all precipitate goes into solution.
- Add 11uL of H20 to a 200uL PCr tube.
- Add 2uL of each digest product to the new PCR tube.
- Add 2uL of 10X T4 DNA Ligase Reaction Buffer to the PCR tube, then add 1uL of the T4 DNA Ligase to the tube.
- Flick the tube to mix contents and spin the tube in a microcentrifuge for a few second to collect the liquid.
- Incubate at room temperature for 10 minutes followed by 80C for 20 minutes. This will deactivate enzymes and improve transformation efficiency.
- The ligation step is now completed. You may wish to store the products at -20C or begin a transformation immediately.
Back To Top
Digestion
- Digestion cuts the DNA at very specific sites with restriction enzymes. This procedure is required for various DNA manipulation techniques including gel electrophoresis and BioBrick assembly. Note: The procedures from this point forward assume that DNA has been amplified and purified. If this is not the case, please read the Polymerase Chain Reaction procedure for amplification and the Mini-Prep procedure for purification.
Materials
- NEBuffer 2
- 10x BSA
- dI H20
- Upstream, Downstream, and Destination Plasmid parts
- Restriction Enzymes (varies depending on digestion)
Procedures
- Begin by thawing the upstream, downstream, and destination plasmid parts along with the NEBuffer 2 and BSA.
- In three separate PCR microcentrifuge tubes labeled upstream, downstream, and destination, add 750ng-1000ng of the respective dried DNA and dilute with dH20 to 38 uL.
- Add 5 uL of NEBuffer 2 and 5 uL of 10x BSA to each tube.
- Add 1 uL of the first appropriate enzyme to each tube. Then add 1 uL of the second appropriate enzyme.
- Flick each tube to mix reagents and incubate at 37C for 1 hour.
- Transfer the tubes to an incubator set at 80C for another 20 minutes. This step will deactivate the restriction enzymes.
- Digestion is now finished and products should be stored at -20C or proceed to Ligation.
Back To Top
Ligation
- Ligation formally joins two or more pieces of DNA together that are already annealed. Note: this procedure requires the products of a successful digestion.
Materials
- DNA Ligase
- 10X T4 DNA Ligase Buffer
- dI H20
- Upstream, Downstream, and Destination Plasmid parts
Procedures
- Thaw the 10X T4 DNA Ligase Reaction Buffer and mix to dissolve the precipitate.
- Add 11 uL of dH2O to a 200 uL PCR tube. Then add 2 uL of each of the digestion products (upstream, downstream and destination) together into this new tube.
- Add 2 uL of 10X T4 DNA Ligase Reaction Buffer to the 200 uL PCR tube.
- Add 1 uL DNA Ligase to the PCR tube and flick to ensure the contents are mized.
- Let the mix stand for 1 hour at room temperature before incubating at 80C for 20 minutes (deactivates enzymes).
- Store the products at -20C until they are needed for a transformation.
Back To Top
Gel Electrophoresis
- Gel Electrophoresis is a technique used to separate out DNA according to its length. DNA is loaded into a well, or hole in an agarose gel, and is then pulled through the gel via electric current (DNA has a negative charge). Smaller DNA fragments travel faster through the gel matrix while larger fragments travel slower. To better determine the length of DNA a ladder is often run parallel to an unknown piece of DNA. The ladder consists of several pieces of DNA of known length which serve as a reference point to the unknown fragment. Note that dyes are important in this procedure since DNA alone is invisible. Specific Dyes are needed to adhered to and travel with the DNA through the gel.
Materials
- Agarose
- 1X TAE Buffer
- Ethidium Bromide
- DNA Ladder
- DNA Samples
- Gel Pouring Dock
- Gel Tray
- Gel Box
- Power Supply
Procedures
- Scale out agarose for gel (0.8% - 2.0%)
- Add agarose powder to appropriately sized glassware
- Add 1X TAE Buffer
- Microwave until boiling
- Allow solution to cool until glassware is comfortable in hand
- Add 2ul ethidium bromide and swirl
- Pour gel solution into level gel tray mounted on the gel pouring dock
- Insert gel comb with appropriate number of wells
- Allow gel to set up for approximately 20 minutes
- Remove gel from pouring dock and submerge just below surface of 1X TAE buffer in gel box
- Carefully remove gel comb
- Load DNA ladder and samples
- Add 3ul of ethidium bromide to the buffer near the positive electrode
- Attach gel box electrodes to power supply and apply desired voltage
- Add another 3ul of ethidium bromide to the buffer near the positive electrode when halfway to completion
- Switch power supply off when gel has run to completion
Back To Top
- Phycocyanobilin (PCB) is a blue phycobilin and is essential for the final testing of our project once implanted into Rhodobacter sphaeroides. It is most easily obtained from Spirulina, a cyanobacteria and popular dietary supplement.
Materials
- Spirulina Powder
- dI water
- Methanol
- HgCl2
- Trifluoroacetic acid (TFA)
- Trichloroacetic acid (TCA)
- Acetonitrile
Procedures
- Rehydrate Spirulina powder in dI water (30ml/g dry weight) for 10 min.
- Centrifuge at 30,000xg for 20 min, decant and save supernatant.
- Precipitate phycocyanin from supernatant with 1% (w/v) TCA by incubation for 1h at 4oC in the dark.
- Collect by centrifugation at 30,000xg for 20 min.
- Wash with methanol (2x 20ml/g Spirulina powder).
- Resuspend blue pellet in methanol (2ml/g powder) containing 1mg/ml HgCl2.
- Incubate for 20h at 42oC in the dark.
- Remove protein by centrifugation at 10,000xg for 10 min.
- Precipitate mercuric ion with 2-Mercaptoethanol (1ul/ml), remove by centrifugation at 30,000xg for 10 min.
- Dilute bilin mixture 10-fold with 0.1% (v/v) TFA.
- Apply to C18 Sep-Pak cartridge.
- Wash cartridge with 0.1% (v/v)TFA (2x 3ml) and acetonitrile/0.1% TFA (20:80; 2x 2ml).
- Elute bilin with 3ml acetonitriel/0.1% TFA (60:40) and dry in vacuo.
- Note:Expected yields are 4 umol PCB per 6g Spirulina
- Thanks to Dr. Clark Lagarias for providing this protocol. ref
Back To Top
Glycerol Stock Solution
- Glycerol Stock Solutions are excellent for preserving and freezing cell cultures. It is very important that cells not be damaged or continue to grow too fast for various parts of our project.
Materials
- Centrifuge/screw top tubes
- 50% glycerol sol
- LB Cultures
Procedures
- Label centrifuge/screw top tubes
- Pipet 300 µl of 50% glycerol solution into each tube
- Pipet 700 µl of LB culture into each tube
- Close & vortex to mix.
- Put in -80 C.
Back To Top
Designing Primers
- We found the Registry's protocol extremely helpful for designing primers and constructed most of our primers from this template. To visit the website, please click [http://partsregistry.org/Help:Primers/Design here].
Back To Top
Colony PCR
- Colony PCR is similar to regular PCR in that it is used to amplify DNA. We have used colony PCR to verify successful transformations by adding grown up cells into the PCR mix, lysing the cells, and then amplifying the DNA. The amplified DNA can then be run through a gel and the number of base pairs can then be compared to an expected result.
Materials
- 0.5uL AccuTaq LA DNA Polymerase
- 5uL 10x Buffer
- 2.5uL dNTP mix
- 1 uL DMSO
- 0.5 uL BioBrick forward Primer
- 0.5 uL BioBrick reverse Primer
- 0.5 uL Enzyme
- 40 uL dI H20
- LB agar plate
- E. coli colony to amplify.
Procedures
- Create the PCR mix by combining the first 8 ingredients.
- Touch the colony of E. coli with a sterile plastic loop and do a small streak across a new plate. Swirl this tip in the PCR mix.
- Begin process of thermocycling with a heat shock of 98C for 30.
- Next begin the cycling steps with 94C for 15 seconds, then 71C for 20 seconds, then 68C for 5 minutes. Repeat these three steps thirty times.
- Move solution to 68C for 20 minutes.
- PCR is now complete and should be held at 4C.
Note: any further streaking should be done with a sterile tip.
Back To Top
Genomic DNA Purification
- Characterization and modeling of individuals parts of our system will require the isolation and purification of genomic DNA from Rhodobacter sphaeroides. One of the main goals of the project is to make BioBricks out of ompR, ompF, pucB/A and the puc promoter coding regions. This DNA however is in chromosomal, not plasmid, form and will require a separate protocol to obtain. Note: This protocol and materials were obtained in a kit called DNeasy from Qiagen. To obtain exact recipes and formulas please contact them.
Materials
- Overnight cultures
- Buffered ATL
- Proteinase K
- Buffered AL
- Ethanol
- DNeasy mini column
- Buffer AW1
- Buffer AW2
- Buffer AE
Procedures
- Harvest cells by taking 1.5mL from overnight cultures and spinning them down to a pellet.
- Resuspend pellets in Buffered ATL
- Add 20uL Proteinase K, mix by vortexing and incubate at 55C for 3 hours shaking every 20-30 minutes
- Vortex 15 seconds and then add 20uL Buffer AL to the samples mixing thoroughly and incubate at 70C for 10 minutes
- Add 200uL ethanol to the sample and mix by vortexing
- Pipet mixture from step 4 into DNeasy mini column sitting in a collection tube. Centrifuge at 12,000RPM for 1 minute and discard the flow through in the collection tube.
- Place the mini column in a new collection tube and add 500 uL of Buffer AW1. Centrifuge at 12,000RPM for 1 minute and discard the flow through and collection tube.
- Place the mini column in a new collection tube and add 500 uL of Buffer AW2. Centrifuge at 12,000RPM for 3 minutes and discard the flow through and collection tube.
- Place the mini column in a clean collection tube and pipet 200 uL Buffer AE on the DNeasy membrane. Incubate at room temperature for 1 minute and then centrifuge for 1 minute at 12,000RPM.
- Repeat elution as described in step 9 with same collection tube to combine both elutes.
Back To Top
- Gel Extraction is intended to recover and reuse DNA that has been purified by gel electrophoresis. The process re-isolates DNA and removes all primers, dyes, and ethidium bromide that may still by present in the band of DNA in the gel, leaving up to 95% of the original DNA. We will be using the GenElute Gel Extraction Kit from Sigma-Aldrich so individual formulas and solutions may be unattainable. Please contact them for more details.
Materials
- gel with desired DNA band
- Column preparation solution
- Gel Solubilization Solution
- Wash Solution Concentrate G
- Elution Solution
- GenElute Binding Column G
- spin columns
- Ethanol
- Isopropanol
- 3M Sodium Acetate Buffer (pH 5.2)
Procedures
- Dilute the Wash Solution Concentrate G with 48mL of ethanol.
- Cut out the desired band of DNA from the gel leaving as little excess gel as possible.
- Weight the excised gel and place into a microcentrifuge tube. Add 3 gel volumes of Gel Solubilization Solution to the tube (for every 100mg of gel, add 300uL of solution). Incubate tube at 60C for 10 minutes or until gel is completely dissolved. Vortex occasionally to ensure gel is evenly dissolved.
- Prepare the binding column by adding 500uL of Column Preparation Solution to the column and centrifuging for 1 minute. Discard the flow through.
- Check the color of the tube containing the dissolved gel. If the solution is red, add 10uL of 3M Sodium Acetate to the mix until the solution turns yellow.
- Add 1 gel volume of 100% isopropanol and mix (may need slightly more isopropanol for gels greater than 2%).
- Load the gel solution into the binding column and centrifuge for 1 minute and discard the flow through.
- Add 700uL of Wash Solution to the column and cetrifuge for 1 minute and discard the flow through. Centrifuge the column again for 1 minute to remove excess ethanol.
- Transfer the column to a new collection tube. Add 50uL of preheated Elution solution (65C) to the column and centrifuge for 1 minute. The desired DNA is now in the collection tube.
Back To Top
Making Competent Cells
- Competent cells are necessary for performing transformations. This protocol prepares cells to take up new, foreign plasmids. Note that different cells may require slightly different preparatory protocols.
Materials
- SOB media
- SOC media
- CCMB80
- Glycerol
Procedures
Preparing Seed Stocks
- Streak cells on an SOB plate and grow for single colonies at 23C. Pick single colonies into 2mL of liquid SOB media and shake overnight at 23C. Add to 15% glycerol. Aliquot in 1mL samples and place in a -80C freezer to store indefinitely. Note competent cells lose efficiently every time they are frozen and then refrozen.
Preparing Competent Cells
- Innoculate 250mL of SOB media with 1mL vial of seed stock and grow at 20C to an Optical Density (OD) of 0.3 for approximately 16 hours.
- Centrifuge at 3,000g at 4C for 10 minutes and discard liquid. Resuspend the cells in 80mL of CCMB80 buffer and incubate on ice for 20 minutes.
- Centrifuge again at 4C at 3,000g for 10 minutes. Discard the solution and resuspend the cells in 10mL of CCMB80 buffer. Test OD of a mixture of 200uL SOC and 50uL of the resuspended cells. Add chilled CCMB80 to yield a final OD of 1.0-1.5.
- Incubate on ice for 20 minutes and then aliquot to 2mL vials and store indefinitely at -80C.
Back To Top
Tissue Flask Experiment
This experiment to monitor the growth and spectra of Rhodobacter shaeroides under anaerobic, photosynthetic conditions was performed as follows:
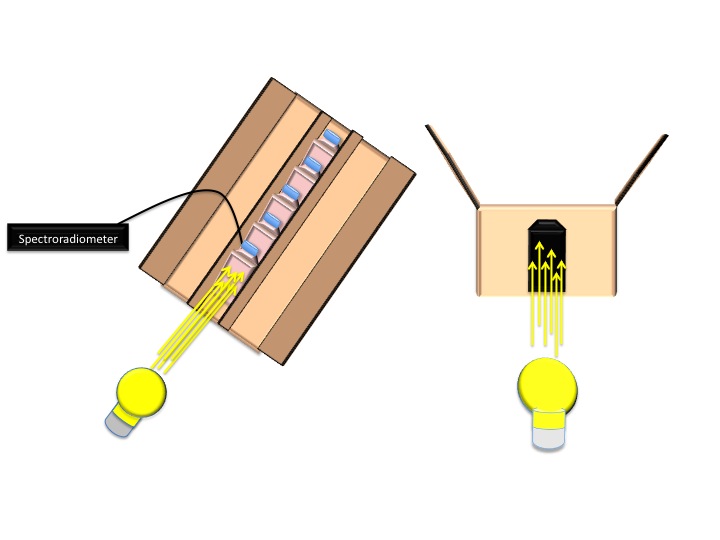
-To ensure optimal growth conditions, an Excella E24 incubator shaker was used to maintain a temperature of 34° C and shaking at 160 rpm and the lid of the incubator was propped open 0.5 cm to prevent overheating. The incubator was covered by a black, opaque cloth to prevent external light from influencing culture growth. Tissue flasks were enclosed by 2 growth boxes inside the incubator, which allowed light to enter only through a tissue flask sized opening on the end nearest the light source. These boxes had the dimensions: 34.8 cm x 18.3cm x 16.8cm.
-5 BD Falcon ™ 50 ml 25 cm² Cell Culture Flasks with blue plug-seal screw caps were used to grow cultures within each box. Cell culture flasks were arranged in series with the first flask recessed 2.5 cm from of box opening and with 2.5 cm separations between each flask. The cell culture flasks were flanked on each side by an opaque barrier so that light incident on each flask was either from the light source, in the case of the 1st flask, or from the light transmittance from the preceding flask in the series
-A constant light source was centered in front of each of the growth boxes The Light source was a 40 watt incandescent light bulb located 10.8 cm from the growth box opening, as measured from the closest point to the box.
-Tissue culture flasks were inoculated with R. sphaeroides cultures in the exponential growth phase from a 50 ml inoculation culture grown at 34° C in the dark in M22 liquid media.
-The test tissue culture flasks were inoculated with Volume = 0.478ml / OD600nm of inoculation culture
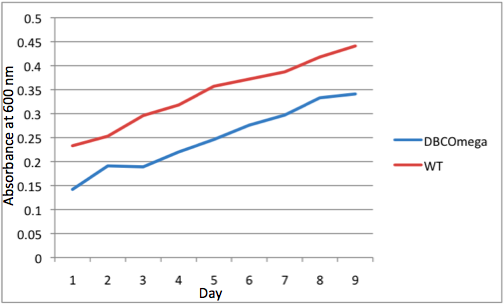
-M22 liquid media was added to flasks for a final volume of 65ml and just after inoculation each flask had OD600nm = 0.011
-Cell culture growth was monitored by measuring OD600nm daily at a standard time for each tissue culture flask. To conduct this measurement,1ml of culture was extracted from each flask for measurement and 1ml M22 liquid media was added to each flask to replace the extracted volume and to ensure minimal oxygen exposure to the culture by reducing headroom.
-How spectroradiometer readings were taken and curves formed (briefly here)


Back To Top
Recipes
Luria Broth (LB) Media
- 10g Tryptone
- 5g Yeast Extract
- 10g NaCl
- 15g Agar
- up to 1 Liter H20
- adjust pH to 7.5
ATCC medium: Van Niel's yeast agar
- 1.0 g K2HPO4
- 0.5 g MgSO4
- 10.0 g yeast extract
- 20.0 g agar
- 1.0 L tap water
- Adjust pH to 7.0-7.2
Super Optimal Broth (SOB)
- 2% w/v bacto-tryptone (20g)
- 0.5% w/v bacto-yeast extract (5g)
- 8.56mM NaCl (0.5g) or 10mM NaCl (0.584g)
- 2.5mM KCl (0.186g)
- ddH2O to 1L
Super Optimal Broth with Catabolite repression (SOC)
- In addition to all of the SOB components,
- 10mM MgCl2 (0.952g)
- 20mM glucose (3.603g)
50X TAE Buffer
- 242 g Tris base (2-amino-2-hydroxymethyl-propane-1,3-diol) (= 2 mole)
- 57.1 ml glacial acetic acid (= 100% acetic acid) (57.19 ml = 1 mole)
- 100 ml 0.5 M Na2 EDTA (pH 8.0)
- ddH2O to 1L
5X TBE Buffer
- 53 g of Tris base (CAS# 37186)
- 27.5 g of boric acid (CAS# 11280)
- 20 ml of 0.5 M EDTA (CAS# 60004) (pH 8.0)
- ddH2O to 1L
Tris EDTA (TE) buffer 5X 1L
- 750 mL d-H20
- 242 g Tris base
- 57.1 mL glacial acetic acid
- 100 mL 0.5M EDTA (93.05 g EDTA in 500 mL d-H20, pH ~8.0)
- fill to 1 L
- adjust pH to 8.5
Agarose Gel (for electrophoresis of DNA >100bp)
- 50 mL 1X TAE buffer
- 0.8 g agarose
- 2.5 microliters EtBr (Caution: EtBr is a known carcinogen)
- Note: Jacob suggested adding 1.0 microliter EtBr to gel and 3.0 microliters to TAE buffer in rig.
CCMB80 Buffer
- 10 mM KOAc pH 7.0 (10 ml of a 1M stock/L)
- 80 mM CaCl2.2H2O (11.8 g/L)
- 20 mM MnCl2.4H2O (4.0 g/L)
- 10 mM MgCl2.6H2O (2.0 g/L)
- 10% glycerol (100 ml/L)
- adjust pH DOWN to 6.4 with 0.1N HCl if necessary
- Note: adjusting pH up will precipitate manganese dioxide from Mn containing solutions.
- sterile filter and store at 4°C
- slight dark precipitate appears not to affect its function
Back To Top
R. sphaeroides Protocols and Recipes
Optimal Growth Conditions
- M22 Media, anaerobic
- 34º C
- Incandescent Light
Recipes
M22 Growth Media
M22 is a Minimal Media that promotes photosynthetic growth. The media is essential for tripaternal mating protocol where it prevents growth of E. coli.
10X Stock: makes up 4 Liters
- Potassium dihydrogen orthophosphate KH2PO4 122.4g
- Diapotassium dihydrogen orthophosphate K2HPO4 120.0g
- DL – Lactic acid (fridge) Na Lactate solution 100.0g
- Ammonium sulphate – big pot (NH4)2SO4 20g
- Sodium Chloride NaCl 20g
- Sodium succinate 173.7g
- Sodium glutamate L – glutamic acid 10.8g
- Aspartic acid DL – aspartic acid 1.6g
- Solution C 800ml
Make up to 2-3 litres, pH to 6.8 and then make up to 4 Litres.
Autoclave as 10 X 500ml flats.
1X M22:
To make up 2 Litres
- 10X stock M22 200ml
- CAA 40ml
- Water 1760ml
Solution C
Makes up to 4 Liters
- Nitrilotriacetic acid 40g
- Magnesium Chloride MgCl2 96g
- Calcium Chloride CaCl2 13.36g
- EDTA 0.5g
- Zinc Chloride (poison) ZnCl2 1.044g
- Ferrous Chloride (poison) FeCl2 1.0g
- Manganous Chloride MnCl2 0.36g
- Ammonium molybdate (NH4)6Mo7O244H2O 0.037g
- Cupric Chloride (poison) CuCl2 0.031g
- Cobaltous nitrate (poison) Co(NO3)2 0.0496g
- Boric acid (orthoboric acid) 0.0228g
Do not autoclave, just freeze at -20°C in 400ml aliquots.
Casamino acids (CAA):
To make 1 Litre
- Casein Hydrosylate acid 50g
Makes up 5% solution to be aliquotted into 200ml.
Batches:
- 1.5 Liters in 2 Liter flasks
- 12 X 80ml in 100ml flasks
- 100 X 10ml in universals.
- For M22 agar add 1.5g agar to 100ml of M22 with no CAA in, store in 300ml flats.
Vitamin Solution
prepare a 10,000 times stock solution of vitamins as follows.
- nicotinic acid 1g
- Thiamine 0.5g
- pABA(p-Aminobenzoic acid) 0.1g
- Biotin (d-Biotin) 0.01g
- Milli-Q water 100ml
Aliquot into 20mls after filter sterilisation.
Add 1ul per every 10ml of M22 media. Do this after you have autoclaved the
media, since the vitamins are labile and the heat will destroy them. When
adding them to melted agar wait until the agar is relatively cool. Freeze the
aliquots you aren't using and keep your working stock in the fridge.
Back To Top
Protocols
Work was done in E. coli and final constructs were transferred to pRKCBC3, which is based on the broad-host range expression plasmid pRK404. This vector contains sequences for conjugal transfer (oriT), replication (oriV). It is maintained at a low copy number (3-4) and features tetracycline resistance (tetR).
Conjugation: Triparental Mating
- Plasmid (pRK404 derived) in E. coli GC5 (or other F- strain)
- Conjugation helper strain: E. coli with pRK2013
- Destination strain: R. sphaeroides DBCΩ(LH2 Knockout with streptomycin resistance)
- Expect to see colonies in 4-6 days
1) Start cultures
- Start 15ml 2 day culture R. sphaeroides DBCΩ with strep 10ug/ml in M22
- Start 15 ml overnight culture E. coli GC5 with plasmid to transfer in LB with strep 5ug/ml tet 5ug/ml
- Start 15ml overnight culture E. coli with pRK2013 in LB with kan 50ug/ml
2) Spin down cultures for 5 minutes
3) Remove supernatant from cultures and resuspend in 1ml antibiotic-free liquid media
- Use LB for E. coli
- Use M22 for R. sphaeroides DBCΩ
4) Combine equal volumes of resuspended cells (~30ul each)
5) Pipette mixture of cells onto well dried M22 plate
6) Allow cells to dry into media for 8hrs at 34º C in the dark
7) Scrape dried cells of M22 plate and streak onto M22 step 5ug/ml tet 5ug/ml
Back To Top




 "
"
