Contents
6-15-2009
- Miniprep of GFP template plasmid, pcDNA5/FRT
| Nr
| pcDNA5/FRT
| GFP
|
| 1
| 4,7
| 39,7
|
| 2
| 5,9
| 14,9
|
| 3
| 3,7
| 3,8
|
| 4
| 5,0
| 7,8
|
- Maxiprep pcDNA/FRT 188.7ng/µL
- Extraction of CMV promoter from 2008 distribution
- Transformation of DH5a cell with CMV promoter
6-16-2009
- DNA synthesis (JeT, cFos, Min)
- Purification of cDNA via 2% / 3% agarosegel
=> Only bands at ca. 50 Bp => no successful amplification
- Site directed mutagenesis of pcDNA5/FRT and mcherry
- Dpn1 digestion
- Transformation of DH5a
6-17-2009
- No transformations could be observed
- Replace Phsuion stocks to Phusion Master Mix from Nathan
- Repeat DNA synthesis with a different protocol: 1:10 - 1:1000 dilution of Oligos, 95° 5', 7* [95° 45 58° 45 72° 1'], add 1:10 diluted primers, 95° 5' 25* same temperatures
- Annealing successful for JeT and Min
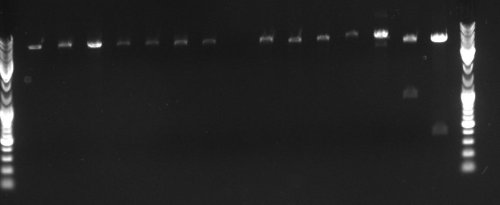
Annealing might not have been successful with Fos beacuse it contains a very repetetive proximal promoter.
[Bild1.jpg|none|frame|Lanes 1-6 Fos (403 Bp); Lanes 7-12 JeT (227 Bp): Lanes 13-18: Min (207 Bp) Lanes 1 : 1µL of 1 :10 diluted oligo 2 : 1 :100 3 : 1 :1000 ;4-6 same, but + DMSO Lanes 7,8,13,14: 1:10 Lanes 9,10,15,16: 1:100 Lanes 11,12,17,18: 1:1000 Even lanes: No DMSO Uneven lanes: DMSO]
- gel purfifcation of JeT and Min
- Repeat PCR of pcDNA5/FRT and mcherry with lower annealing temperature (58°C) and fresh Polymerase: Follow PCR protcol from Strategene site directed mutagenesis kit.
mcherry PCR might not have been successful because
we used a template we didn't make ourselves - does it contain TE?
- PCR worked for pcDNA5FRT but not for mcherry
- DpnI digest of pcDNA5FRT 1,3,4,6
6-18-2009
- DNA synthesis for Fos (proximal) and Fos (core)
 Left two lanes: Proximal (repetetive), right two lanes (core) - Transformation of DH5a with pcDNA/FRT ΔPstI
Gave many 1000 colonies. Probably a contamination (?), whci hwould explain why there was no mutagenesis (see below)
- HeLa cells, MCS7 and U20S were splitted 1:3. Therefore DEMED medium (containing additionally 10% FCS, Pen-Strep, glutamate and non-essential aminoacids) was removed. Cells were washed afterwards with HBSS and trypsinised with 1 ml of trypsin and incubated for 5 min. at 37°C 5% CO2. The used flasks were filled up to a final volume of 7 ml with DMEM. 2 ml of the suspension was transfered to a new flask, 5 ml DMEM was added and incubated for another 3-4 days at 37°C and 5% CO2.
6-22-2009
- Split cells (Split again Thursday!)
- Prepare 6well-plate with U20S cells for Zeomycine assay
- Miniprep pcDNA/FRT ΔPstI
- Digest with PstI => No mutation
- Use Stratagene QuickChange XL kit for mutagenesis PCR (mcherry, pcDNAand Supercompetent Gold TOP10 cells for transformation => reasonable amount of colonies
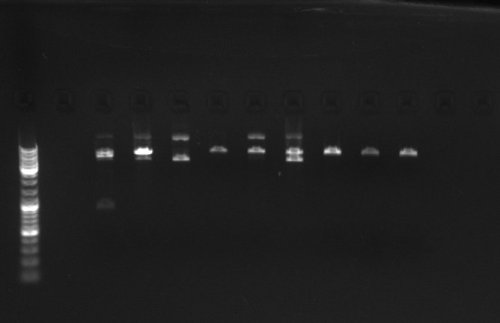
6-23-2009
 2: pcDNA S-1; 3: pcDNA S-4; 4: pcDNA S-2; 5: pcDNA S-3 6:pcDNA S-5 7: pcDNA S-6 8: pcDNA S-7 9: pcDNA S-8; 10: mCherry D-1 11: mcherry S-2; 12: mcherry S-3; 13: pcDNA D-1 14: pcDNA D-2 15: pcDNA control 16: mcherry control - Added Zeomycine to 6-well platees
- Miniprep mutagenisis PCR
- Digest with PstI => Mutagenesis successful
- PCR to remove EcoRI from pcDNA5/FRT using Phusion polymerase
6-24-2009
- DpnI digest
- Transformation of Supercompetent Gold TOP10 cells with pcDNA5 ΔEcoRI ΔPstI
6-25-2009
Split cells!
- Observation Zeomycine: 0 no killing, 50, 100: significant amount of cells death. 250: ca. 30% of cells death. 500, 750: At least 50% death.
- Site directed mutagenesis to remove EcoRI site with stratgene kit, as no colonies had formed previously.
6-26-2009
Change 6well plate medium!
- Observation: 0 Zeomycine 100% confluent; 50, 100: some death cells; 250: Many death cells; 500: high confluency (was higher originally), many death cells; 750: most cells death
- Transformation with pcDNA5/FRT ΔPstI EcoRI removal PCR product successful (~ 300 colonies)
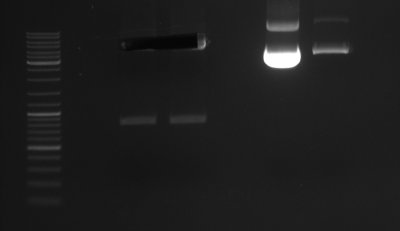
- Miniprep, Test digest with EcoRI
 Lane 1: Ladder; Lane 3: Control; Lane 4-12: Clones of site directed mutagenesis to remove EcoRI 6-29-2009
- Observation Zeomycine assay: 0 Zeomycine some death, 100% confluent; 50, 100: some remaining cells alive; 250: All cells death but in some areas; 500, 750: All cells death
- Abort Zeomycine assay. Rasult: Use 250-500 µg/µl
- Maxiprep of pcDNA5/FRT ΔPstI ΔEcoRI
- Digest pcDNA5/FRT with PstI and MfeI, digest synthesized promoters with the same enzymes
- Digest pcDNA with BclI -> BclI does not cut -> cloning strategy must be changed to use ApaI instead of BclI (new primers designed and ordered)
- Gel purification of vector backbone
 Lane 2+3: Preparation of Vector backbone after MfeI/PstI digest; Lane 5: plasmid (pcDNA5/FRT ...) Lane 6: BclI digest of plasmid (same behaviour as Lane 5) - Ligation of pcDNA5/FRT ΔPstI ΔEcoRI -MCS with JeT and Min promoters
6-30-2009
- Many colonies on all transformations
- Pick colonies
- Prepare media
- PCR of mcherry, GFP
7-01-2009
- Miniprep of pcDNA5/FRT ΔPstI ΔEcoRI JeT, pcDNA5/FRT ΔPstI ΔEcoRI Min, pcDNA5/FRT ΔPstI ΔEcoRI (all unmethylated)
- Analytic digest with MfeI and PstI to retrieve insert => contaminations and uncut (original, removed insert shows up again), possibly one positive result
- Analytic digest of the positive result with SpeI and NdeI to get a larger insert
=> ligations had not worked
- mcherry PCR worked, GFP didn't
7-02-2009
- Digest pcDNA5/FRT ΔPstI ΔEcoRI from -dam strains with BclI and PstI, Same for mcherry PCR
- PCR purification
- Ligation
- Transformation
7-03-2009
- Colony PCR - Colonies 1,2,3,8,10 are promising (to be picked, mini-prepped and test-digested)
- Transformation of DH5a with Part BBa_E0040 (GFP)
7-04-2009
- Inoculation of LB-Amp media with colony 1,2,3,7,8 and 9
- Incubation over night at 37°C
7-05-2009
- Miniprep of overnight culture
- Digestion of pcDNA5/FRT ΔPstI ΔEcoRI - mCherry ligation with Pst1 (1h 37°C)and Bcl1 (1h 50°C)
- HeLa cells, MCS7 and U20S were splitted 1:5 (see above)
- 6-well plates with the human cell lines were started for transfection (~10^5 cells per well)
- Agerose Gelelktrophorese
 (lane 1 marker; lane 2 control (undigested); lane 3 colony 1; lane 4 colony 2, lane 5 colony 3; lane 6 colony 7/1; lane 7 colony 7/2; lane 8 colony 8; lane 9 colony 9 7-06-2009
- Digest pcDNA5/FRT ΔPstI ΔEcoRI mcherry(PstI-BclI), JeT and Edelman/Min with PstI and MfeI
- SAP digest, gel purification of backbones
- Ligation
- Transformation
- Transfect HeLa, U20s and MCS7 cells with pROG4 (??) for stable integration of a FRT site
7-07-2009
- HeLa cells, MCS7 and U20S were splitted 1:5 (see above)
- Preparation of 6well-plate with HeLa cells, MCS7 and U20S for transfection (stabel integration). Therefore 1*10^5 cells were deluted with DMEM medium to a final volume of 2.5 ml.
- Preparation of 8well-plate with Hela cells for transfection (pcDNA5/FRT-mCherry-promoter-ligation). Therefore 1.5*10^4 cells were deluted with DMEM to a final volume of 0,4 ml.
- Mini-prep of GFP
- PCR of GFP (O. 47 and O.48) (checked via gel)
- Inoculation of medium with colonies (pcDNA5/FRT ΔPstI ΔEcoRI mcherry-min-ligation / pcDNA5/FRT ΔPstI ΔEcoRI mcherry-JeT-ligation)
7-08-2009
- Digest P.8 (GFPb3mut) PCR (O.47 and O.48) product with Bcl and PstI
- Digest P.7 (unmethylated) with BclI and PstI
- Ligation
- Transformation
- Test digest of pcDNA5/FRT ΔPstI ΔEcoRI mcherry-min-ligation / Jet-ligation -> ligation worked (checked by gel)
7-09-2009
- Randomized promoter synthesis of HIF-1 using
L1389.O53 HIF-1 a 4µL
L1389.O54 HIF-1 b 4µL
L1389.O55 Ap1 0,5µL
L1389.O56 Random 9 µL
L1389.O57 Sp1 0,5 µL
L1389.O58 Stop3' 1 µL
L1389.O59 Stop5' 1 µL
To 200µL; dilute mix 1:10
7 cycles as described for gene synthesis (+/- DMSO), then add primers (O.58, O.59) 1:4 and 1:10 diluted
- Digestion of P.9 with ScaI
- Transfection with P.9 for stable integration; Transfection with mcherry - Jet/min ligation (transient)
 From left to right: 2* no DMSO , 2* DMSO; 1:4 1:10 1:4 1:10 Notice that indeed this is no smear but individual bands.
==
7-23-2009
- Retrace and comprehend last weeks' doings
- Used pSB1A3 with mCherry (2009 spring distribution plate 1; well 1K) for construction of submission plasmid (add 15 µl water to well and mix)
- Transformed DH5α cells once with pSB1A3 and once with plasmid 6 (pcDNA5/FRT ΔPstI ΔEcoRI)
==
7-24-2009
- start new cell culture (new media, new cells (HeLa, U2OS from Michaela)...)
* glutamine and trypsin aliquots are now in freezer II (drawer #3)
- pick transformed cells (had single colonies with plasmid 6 as well as with pSB1A3) and culture during the day (5 ml in 37°C incubator with rotation)
- synthesize I_pSB1A3 and I_pcDNA from oligos (I_pSB1A3 and I_pcDNA contain BBb standard) twice (first try didn't work)
- maxi (250 ml w/ 250 µl Amp and 2,5 ml pre-culture) --> incubate over night
* those inserts and the according vector will be digested with SspI and PciI (pSB1A3) or SspI and SphI (plasmid 6) respectively
7-27-2009
- PCR of EGFP with BclI/PstI primers
- Digest P.6 with SspI and SphI, P.23 with SspI and PciI
- Digest I_pcDNA with SspI and SphI
- Digest I_psb1A3 with SspI and PciI
- Ligate
- Split cells (HeLa, U2OS --> 1:10)
7-28-2009
- Gel electrophoresis of ligation (plasmid 6 + I_pcDNA separate, plasmid 6 + I_pcDNA ligated, pSB1A3 + I_pSB1A3 separated, pSB1A3 + I_pSB1A3 ligated)
[Image:072809_P6_insert_pSB1A3_insert.jpg|none|frame| From left to right: plasmid 6 + I_pcDNA separate, plasmid 6 + I_pcDNA ligated, pSB1A3 + I_pSB1A3 separated (not visible here), pSB1A3 + I_pSB1A3 ligated, 1kb DNA Ladder]
- Purification of ligated plasmids with inserts from gel
- transformation of the two ligated plasmids (plasmid 6 + I_pcDNA and pSB1A3 + I_pSB1A3), pFRT/lacZeo, mGFPc1, mGFPn2
- mGFPc1 and mGFPn2 are two plasmids from Yara containing eGFP
- transient transfection of HeLa cells with 3J2, 8J4 (plasmid w/ mCherry and synthetic Jet-Promotor), 8M1, 8M3 (plasmid w/ mCherry and synthetic Min-Promotor), 3, 8 (plasmids with mCherry and CMV-Promotor), positive and negative control (mGFPn1 or plasmid 6 respectively)
- Lab Tek 8 chamber allocation:
| 3J2
| 8M1
| 8M3
| 8J4
|
| 3
| 8
| neg
| pos
|
- made fresh LB-medium and LB-amp plates
7-29-2009
- Transformation of plasmid 6 + I_pcDNA, pSB1A3 + I_pSB1A3 did not work
- Gel purification of digested plasmid6 and I_pSB1A3 to delete old Insert
- New ligase reaction of plasmid6 + I_pcDNA, pSB1A3 + I_pSB1A3, with higher DNA concentrations
- Gel purification of eGFP n1 PCR
- Transformation of 8M1, 8M3, 8J2, 8M4 in SCS110 supercompetent cells, and plasmid6 with I_pcDNA, pSB1A3 with I_pSB1A3 in DH5α cells
- Plating of Transformed cells
- Overnight culture of DH5α with pFRT/lacZeo
- sequence of Jet and mCherry in integration plasmid arrived --> mCherry mutagenesis did not work --> only half of mCherry is in the plasmid, therefore no fluorescence after transient transfection
7-30-2009
- Over day culture of 8M1, 8M3, 8J4, 3J2 in SCS non-methylating cells
- transformation of plasmid6 with I_pcDNA, pSB1A3 with I_pSB1A3 in DH5α cells did not work, maybe mistake in ligation step
- synthesis of I_pSB1A3 and I_pCDNA in two parts for each (idea: maybe previously synthesized inserts (I_pSB1A3 and I_pcDNA) were too long --> synthesize two shorter parts, digest them with EcoRI and ligate them)
- mutagenesis of mCherry at PstI location
- PCR of previously (?) mutagenized mCherry
- Digest I_pSB1A3 with SspI, PciI and EcoRI
- Digest pSB1A3 with SspI, PciI
- Digest I_pcDNA5FRT with SspI, SphI and EcoRI
- Digest pcDNA5FRT with SspI, SphI
- Digest mcherry PCR, EGFP PCR with PstI, BclI
- Miniprep of 8M1, 8M3, 8J4, 3J2
- Digest 8M3 and 3J2 with PstI, BclI
- Ligate I_x with x and 8M3 / 3J2 with mcherry/GFP
cell culture:
- transient transfection:
- no fluorescence with 8M1, 8M3, 8J2, 8M4 (makes sense, because we had it sequenced and there's only half of mCherry in there)
- positive control worked (GFP)
- stable transfection:
- removed old medium
- added fresh medium with Zeocin (stock at 100 mM (used all the stock for 50 ml medium) --> add to medium to get a concentration of 200 µM (1:500))
IDs created
- 30.7.1 = 3J2 - mcherry fragment + GFP
- 30.7.2 = 8M3 - mcherry fragment + GFP
- 30.7.3 = 3J2 - mcherry fragment + mcherry
- 30.7.5 = BBb integration plasmid (pcDNA5/FRT derived)
- 30.7.6 = BBb submission plasmid (pSB1A derived)
- 30.7.7 = 8M3 - mcherry fragment + mcherry
7-31-2009
- yesterdays transformations did not seem to work (no colonies on any plates) - at 3pm, colonies were found
- Redo Restriction, ligation of 8M3, 3J2 with GFP (31.7.1, 31.7.2)
- Transformation of 30.7.5 and 30.7.6 (pSB1A3 + I_pSB1A3, p6 + I_pCDNA)in SCS110 supercompetent cells
- PCR of RFP from pSB1A3 (registry spring distribution 2009 plate 1 well 1K) with primers containing BBb (inner restriction sites --> SpeI, NheI)
- same for ccdB ("death gene" from registry, spring distribution 2009 plate 1 well 3K)
cell culture:
- HeLa and MCF-7 were relatively dense --> splitted 1:10 (so they can be splitted again on Monday and transfection can be prepared)
- U2OS were not quite as dense --> splitted 1:6
IDs created
- 31.7.1 = Ligation of 3J2 + GFP (transformation tbd)
- 31.7.2 = Ligation of 8M3 + GFP (transformation tbd)
8-01-2009
- Transform DH5a with 31.7.1, 31.7.2
8-02-2009
- Start Overnight cultures with 30.7.1, 30.7.2, 30.7.3, 30.7.5, 30.7.6, 30.7.7, 31.7.1, 31.7.2
8-03-2009
- Miniprep of 30.7.1, 30.7.2, 30.7.3, 30.7.5, 30.7.6, 30.7.7, 31.7.1, 31.7.2
- Analytic digest with SphI/PstI for everything but 30.7.5, 30.7.6
- Analytic digest with SspI/NheI for 30.7.5, 30.7.6
- Gel, sequencing of pcDNA plasmids w/ Jet or Min and mCherry or GFP (test plasmid; also to be transformed tomorrow)
 From left to right: 30.7.7#1-4 (used 30.7.7#2 as p.12); 31.7.1#1-4 (used 31.7.1#4 as p.30); 31.7.2#1-2; ladder; control (8M3, 3J2); 31.7.1#3,5-8 (used#6 as p.31); 30.7.1#1-4; 30.7.2#1  From left to right: 30.7.2#2-4; 30.7.3#1-3; ladder; 8m3, 3j2 (control); pcdna5+1#1; psb1a3+I #1,2; pcdna5/FRT (control), psB1a3 - insert synthesis for BBb-standardizing of submission and integration plasmids
- overnight culture of P.30, P.31 and P.13 (???)
8-04-2009
synthetic promoters
- Sequance analysis (alignement to be uploadad)
- Maxiprep p12, p30, p31
BBb submission plasmid
- Digestion of vectors
- Gel purification of vectors
- PCR purification of overnight insert ligation
- Start overnight ligations
8-05-2009
- over night culture (250 ml) of DH5alpha for new competent cells
synthetic promoters
- Re-do insertions of Min and JeT with correctly synthesized promoters
BBb submission plasmid
- Transformations of ligations
8-11-2009
- 1. PCR-amplification of Jet promoter with primer 106 and 71 and template p31
- 2. PCR-amplification of eGFP with primer 97 and 98 and template peGFP-N1
- (PCR-amplification of CMV-promoter with primer 109 and 108 and template pGFP-N1 => failed => wrong primers)
- (PCR-amplification of CMV-promoter with primer 109 and 110 and template pGFP-N1 => failed => wrong primers)
- (PCR-amplification of CMV-promoter + eGFP with primer 109 and 98 and template 3J2 => failed => wrong primers)
- 3. PCR-amplification of CMV-promoter with primer 109 and 108 and template p6
- 4. PCR-amplification of CMV-promoter with primer 109 and 110 and template p6
- PCR purification of all PCR-product
- Restriction digest of peGFP-N1 and Jet PCR-product with AseI and PstI (NE-buffer3 + BSA, 1h at 37°C)
- Restriction digest of p7 plasmid and eGFP-PCR-product with BCl1 and Pst1 (NE-buffer3 + BSA, 0,5h at 37°C and 0,5h at 50°C)
- Gelelectrophoresis of restricted plasmids and PCR-Products (1% Agerose gel)
- Gelextraction of backbones
- Ligation of Jet with peGFP-N1-backbone (molar ratio 3:1)
- Ligation of eGFP with p7-backbone (molar ratio 3:1)
8-12-2009
- PCR-amplification of CMV-promoter with primer 109 and 108 and template pcDNA5/FRT => failed => wrong primers
- PCR-amplification of CMV-promoter with primer 109 and 110 and template pcDNA5/FRT => failed => wrong primers
- Cloning into p31, Transformation
- Pick colonies of the previous day's successful transformations (p.23 + BBB_JeT)
8-13-2009
- Minipreped 8 clones of pSB1A3 w/ BBbed Jet
- overday culture (5 ml with amp/kana) of 1,3,4,5 from 8-11-2009
- Repeat cloning of JeT into pEGFP-N1 using AseI and PstI
8-14-2009
Promotors
- minipreped 3-5 (from 8-13-2009)
- control digest of 3 (Nhe1, Spe1), 4 (Spe1, Hind3), 5 (Pst1, Sph1), pSB1A3 (Spe1)
- gel electrophoresis of control digests and uncut Plasmids 3-5, pSB1A3
8-15-2009
- checked transfected cells of flourescence: both transfections with jet/EGFP in pCDNA5/FRT worked (green flourescence), positive controle worked
8-16-2009
- picked another 8 colonies from plates 3-5 (see 8-11-09) and started overnight culture
8-17-2009
- Synthesized HIF: 2µL LV_HIF-1 a 2µL LV_HIF-1 b 1µL LV_CREB 0.1µL each LV_Ap1 LV_Sp1 1.5µL each LV_Stop3, LV_Stop5; 4.5 µL LV_Random
- Synthesized p53: 4µL p530.1µL each LV_Ap1 LV_Sp1 LV_CREB 1.5µL each LV_Stop3, LV_Stop5; 4.5 µL LV_Random
- Synthesized negative control: 4.5µL LV_empty 1.5µL each LV_Stop3, LV_Stop5; 4.5 µL LV_Rando
- Yielded product in the range of 5kb-200bp
- Cut three fragment (long, medium short); PCR purification
8-18-2009
Cloning of cmv core promoter in Jet GFP plasmid (p31)
- PCR of cmv core promoter from plasmid 6 whole volume 50 µl: 25 µl Phusion MasterMix, 1 µl 1:4 diluted forward primer, 1 µl 1:4 diluted reverse primer, 0.5 µl p6 plasmid maxiprep as template, 22.5 µl DNase free water.
- PCR program: 98 ˚C 30s || 98˚C 10s | 60 ˚C 20s | 72 ˚C 20s || 72 ˚C 5 min | 4 ˚C forever amplied 30 cycles.
- PCR product was purified with PCR purification kit and then all digested with HindIII and NheI-HF in Buffer 4 + BSA for 1 hour. Vector (p31) (5 µl maxiprep product) was also digested with HindIII and NheI-HF for 2 hours. for each digestion 0.5 µl enzyme each.
- Digestion products were gel purified and ligated with volume ratio of 2:1 (Vector : insert) ligation (10 µl): 2µl HindIII and NheI-HF digested p31 vector, 1 µl
HindIII and NheI-HF digested PCR product (CMV core promoter from p6), 1 µl ligation buffer (Fermentas), 1 µl T4Ligase, 5 µl water. ligation was done at room temperature for 1 hour.
- standard transformation in DH5 alpha plated on amp plate.
- clony PCR on next day verify the positive colonies.
8-19-2009
- PCR of BBbJeT from plasmid P.31 with O.70 and O.71 (correct BBb prefix/suffix oligos)
- digested PSB1A3 from registry with EcoRI and PstI
- then heat inactivation, SAP digest and gel extraction
- digested PCR product of BBbJeT with EcoRI and PstI and subsequent PCR purification
- ligation over night at 16°C (ligation product: p.49 --> pSB1A3 with correct BBb standard)
8-20-2009
- transformation of ligation (p.49)
8-21-2009
- minipreped PSB1A3_BBbJeT (P.49)
- test digested with NheI and SpeI
- works --> sequenced!
8-24-2009
- 72 bp missing in PSB1A3 backbone!
- sequenced PSB1A3 from registry --> also missing bp --> e-mailed registry people
- PCR of P.49 (PSB1A3_BBbJeT) with O.72 and O.87
- digested P.6 with SspI and SphI
- then heat inactivation, SAP digest and gel extraction
- digested PCR product (long insert with all the standard stuff from pSB1A3) also with SspI and SphI
- ligation overnight at 16°C (ligation product: p.51 --> standardized pcDNA5/FRT plasmid)
8-25-2009
- use BBbJet PCR product (8-19-2009; with complete BBb standard, including NotI restriction site) and digest with EcoRI and PstI; then PCR purification
- digested P.31 (pcDNA5/FRT with JeT) with EcoRI and PstI
- PCR of CMV (from P.6) with O.108 and O.109
- transformed PSB1A3 with ccdB (iGEM 2009 spring submission plate 1 well 3K) into ccdB surviving cells (invitrogen)
8-26-2009
- started maxi prep of PSB1A3_ccdB
- digested CMV PCR product with NheI and SpeI --> gel purification
- digested P.51 with NheI and SpeI --> gel purification
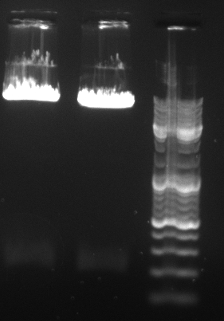
- ligated correct BBbJet into P.31 and transformed
- ligated BBb_CMV into P.51 and transformed
- transformed p.51 (colony 1)
- Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
| HIF
| NFkB
| p53
| constitutive
| empty
|
2,5µL HIF-1 (O.53)
2,5µL HIF-2 (O.54)
1µL CREB(O.89)
3µL HIF-rev (O.189)
3µL Random (O.56)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
.2µL each Ap1, Sp1 (O.55, O.57)
| 2,5µL NFkB-1 (O.93)
2,5µL NFkB-2 (O.94)
3µL NFkB-rev (O.194)
3µL Random (O.56)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
.2µL each Ap1, Sp1, CREB, NFY (O.55, O.57, O.89, O.90)
| 5µL p53 (O.91)
3µL p53 rev (O.188)
3µL Random (O.56)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
.2µL each Ap1, Sp1, CREB, NFY (O.55, O.57, O.89, O.90)
| 2µL Sp1 (O.57)
1µL Ap1 (O.55)
1µL CREB (O.89)
0.5+0.5µL NFkB (O.93/94)
4µL Random (O.56)
1µL NFkB rev (O.194)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
| 5µL Random (O.56)
5µL Empty (O.95)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
|
- Run Gene synthesis PCR (compare material and methods)
- Gel purify: Exclude everything shorter than 100 bp. Excise two fragments: Everything < 750bp, and everything >750bp
 Preparative (2% agarose) gel of random promoter synthesis. PCR product length is spread between >6000 and <100 bp. 8-27-2009
- PCR of mCherry from p.41_muta with O.50 and O.71
- digested PCR product with AvrII and BclI and gel purified product (to be the insert)
- digested p.7 with NheI and BclI --> looks strange (three bands) --> redo
- digested p.7 with NheI, then PCR purification
- PCR of p.29 (pSB1A3 with P1010) and p.23 (pSB1A3 with J04450) with O.176/O.177 or O.174/O.175 respectively
- digested PCR products with EcoRI and PstI
- gel purification of pcr product
- digested p. 49 with EcoRI and PstI
- gel extraction of plasmid backbone of p.49 (pSB1A3 with BBb-standard flanking regions)
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
- Cut gel extracted promoters with SpeI/HindIII, Cut p.31 with SpeI/HinDIII. Use a long digestion time (1,5h + 1h SAP)
- Gel purify vector, PCR purify inserts
- Ligation: 2,5h @ 20°C
- Transformation
---
Protocol optimization
- In order to find out the otpimal conditions: Run a gradient PCR for HIF and const. synthesis (using oligo concentration as specified on 8-26-2009):
|
| 1
| 2
| 3
| 4
| 5
|
| Const, PCR purification after 1st 7 cycles
| Sample 1: 57°C 1st annealing
58°C 2nd annealing
| 2: 58,36°C 1st
61,82°C 2nd
| 3: 59,27°C 1st
64,36°C 2nd
| 4: 60,64°C 1st
68,18°C 2nd
| 5: 62°C 1st
72°C 2nd
|
| HIF, PCR purification after 7 cycles
| Sample 6: 57°C 1st annealing
58°C 2nd annealing
| 7: 58,36°C 1st
61,82°C 2nd
| 8: 59,27°C 1st
64,36°C 2nd
| 9: 60,64°C 1st
68,18°C 2nd
| 10: 62°C 1st
72°C 2nd
|
| Const, no PCR purification after 7 cycles
| Sample 11: 57°C 1st annealing
58°C 2nd annealing
| 12: 58,36°C 1st
61,82°C 2nd
| 13: 59,27°C 1st
64,36°C 2nd
| 14: 60,64°C 1st
68,18°C 2nd
| 15: 62°C 1st
72°C 2nd
|
| HIF, no PCR purification after 7 cycles
| Sample 16: 57°C 1st annealing
58°C 2nd annealing
| 17: 58,36°C 1st
61,82°C 2nd
| 18: 59,27°C 1st
64,36°C 2nd
| 19: 60,64°C 1st
68,18°C 2nd
| 20: 62°C 1st
72°C 2nd
|
8-28-2009
Protocol optimization
- Gel extraction of yesterday's gradient PCRs (one fragment per lane, include everything > 100 Bp)
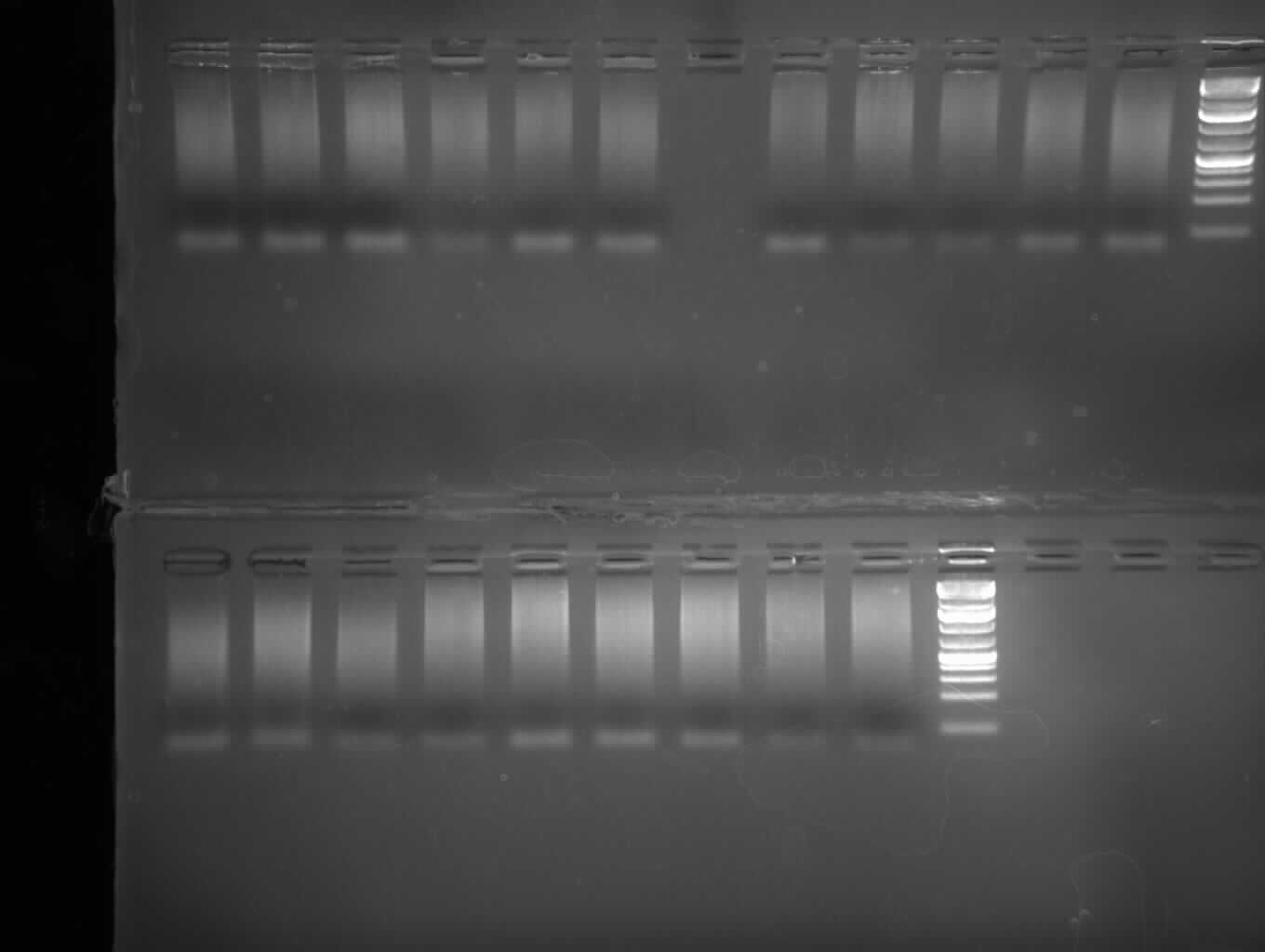 Upper Gel:Lane 1-5: Sample 1-5; Lane 8-10: Sample 8,9,10 (Sample 6 and 7 were lost); lane 11: Sample 11 Lower Gel: Lane 1-4: Sample 12-15; Lane 5-8: Sample 16-20 - miniprep of P.51 (colony 1), P.51 with BBb_CMV and P.31 with complete BBb_Jet
- test digested P.31 with complete BBb_Jet with NotI
- test digested P. 51 and P.51 with BBb_CMV with NheI and SpeI
- digested P.7 that was already digested with NheI with BclI (50°C) for 2,5 hours, because it didn't work well yesterday --> put on gel
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
- Pick 25 colonies each from plates containing putative long and short p53,NFkB,HIF promoters; Pick 15 colonies each from plates containing long and short putative constitutive promoters; Pick 6 colonies each from plates containing long and short random promoters.
- important plasmids (finished):
- p.10: pcDNA5/FRT/eGFP (with original CMV = positive control for transfection)
- p.31: pcDNA5/FRT/eGFP_BBbJeT (complete BBb standard now)
- p.48: pcDNA5/FRT/eGFP_BBbJeT_CMV (incomplete BBb standard)
- p.49: pSB1A2_BBbJeT
- p.51: pcDNA5/FRT_BBbJeT (with standard flanking regions from pSB1A2)
- p.53: pcDNA5/FRT_BBbCMV
8-29-2009
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
- Miniprepped the following colonies:
- HIF S 1-24
- HIF L 1-24
- NFkB S 1-24
- NFkB L 1-24
- p53 S 1-24
- p53 L 1-24
- Const S 1-12
- Const L 1-12
- Empty S 1-4
- Empty L 1-6
8-31-2009
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
- Test digest, nanodropping
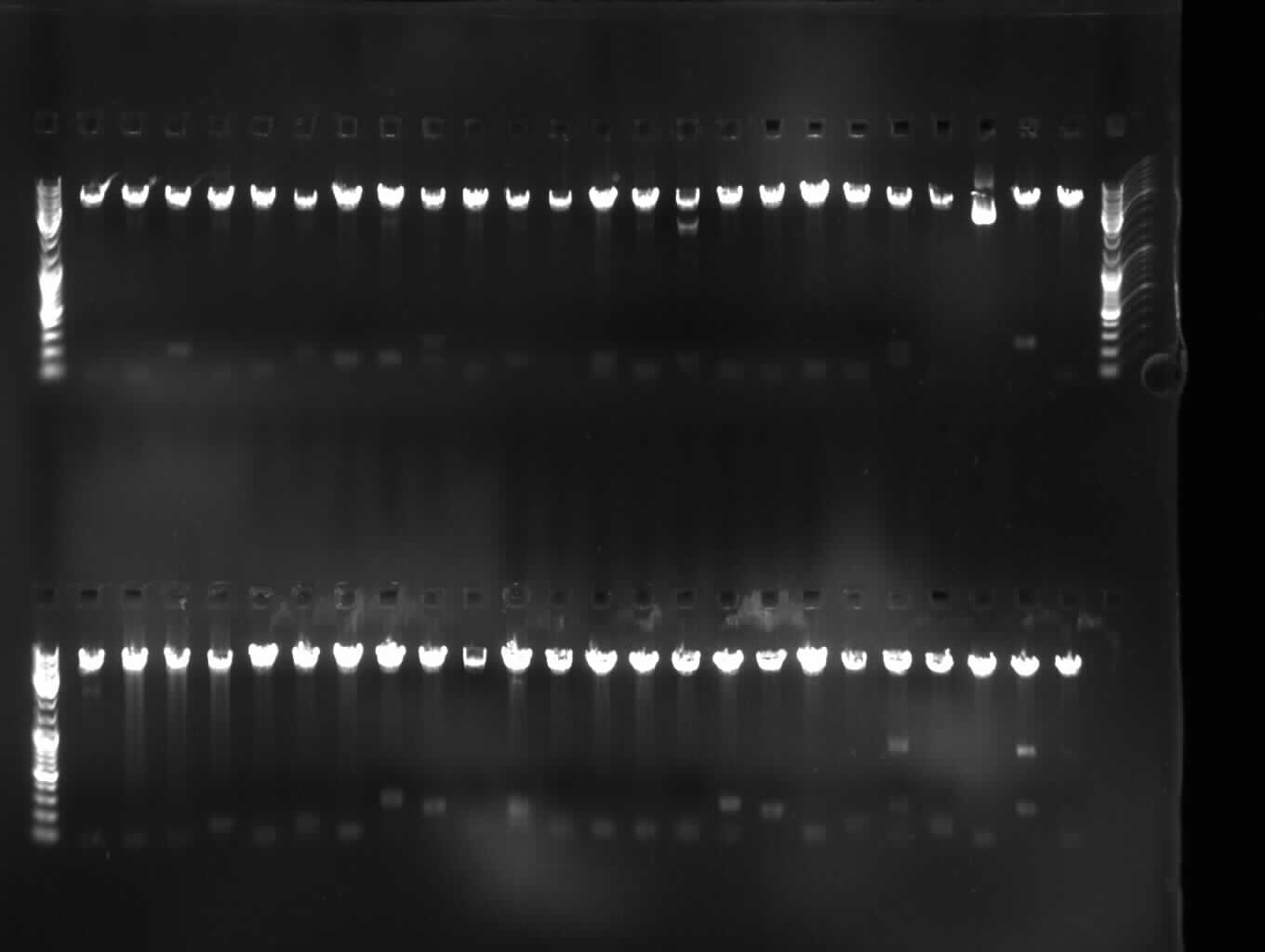 Upper Gel:HIF S Lower Gel: HIF L |  Upper Gel:NFkB S Lower Gel: NFkB L |
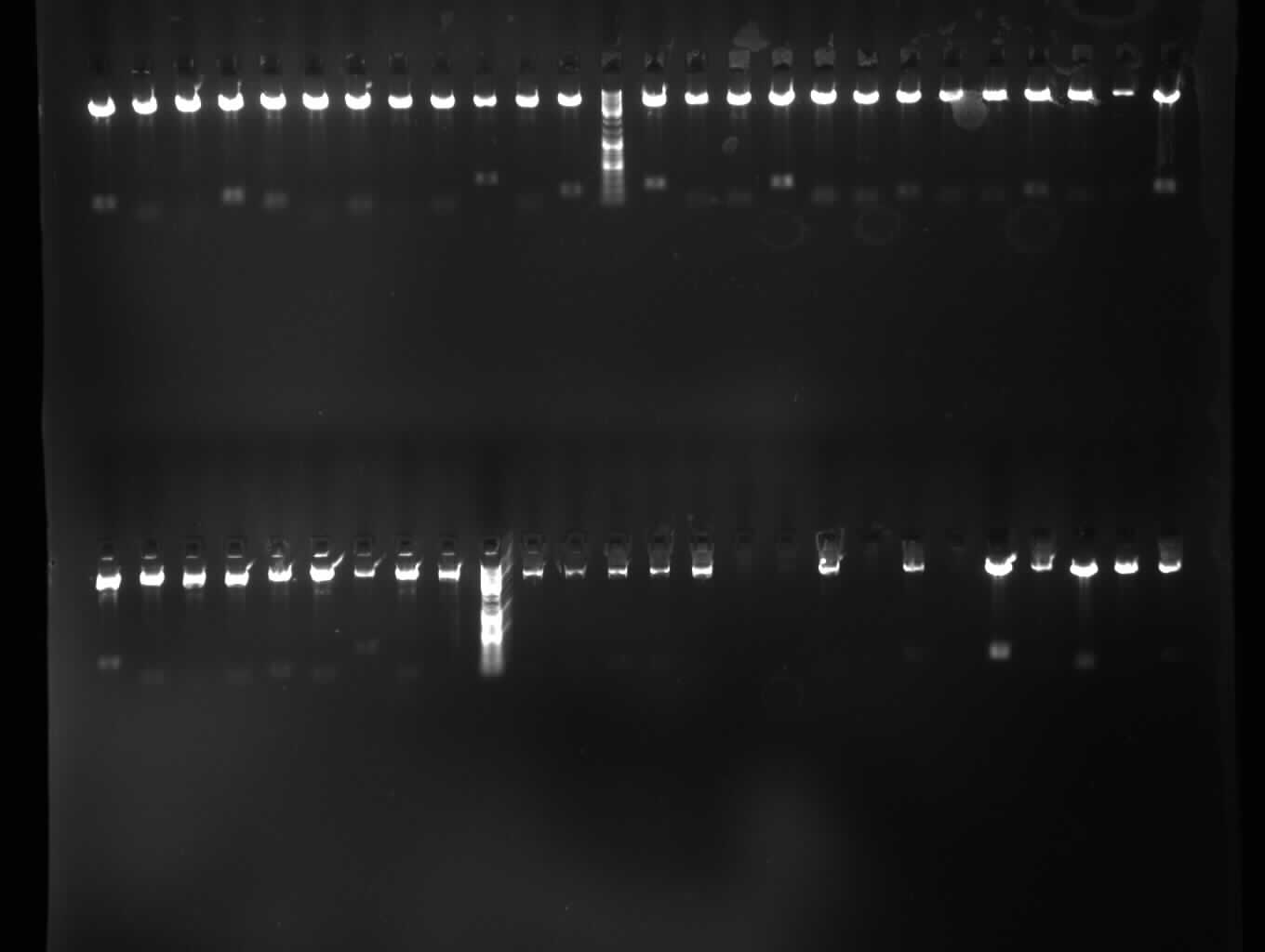 Upper Gel:const S, ladder, const L Lower Gel: Empty S 1-4, Empty L 1-6, p53 1-16 |
|
- Picked DNAs to be transfected and diluted to 33,33 ng/µL:
- HIF S 3-20,23,24
- HIF L 4,6-9,11,16,17,19,20,21,23
- NFkB S 2,4,5,7,15-18,23
- NFkB L 9,14,16,22-24
- p53 all
- const S 4,5,10,12
- const L 1,4,5
- empty all
9-01-2009
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter:
- Induction and prelimary screening
- HIF was induced by hypoxia bag for 1,5 h
- p53 was induced by topoisomerase inhibitor
- NFkB was induced by TNF-a
Protocol optimization
- Cloned optimization samples 1-5 into p31 (ligations, transformations)
- Tried the following PCR protocol for HIF (same mixture as above) in order to increase construct length (Sample thus generated is referred to as Sample W below):
| | |
|
| Initiale denaturation | 95 °C, 5 min | 1 cycle
|
|
|
| denaturation/annealing | 95 - 58 °C decrement step, 15 sec per degree Celsius
|
| annealing | 58 °C, 20 sec
|
| extension | 72 °C, 1 minute | 7 cycles
|
|
|
| | 4 °C | forever
|
|
| PCR purification
|
|
| 25 cycles as normal (69°C annealing temperature)
|
9-02-2009
Synthesis of HIF, p53, NFkB responsive promoters; constitutively active promoter; empty/random promoter: Screening results
- All constitutive promoters were active
- Random promoters were non-active
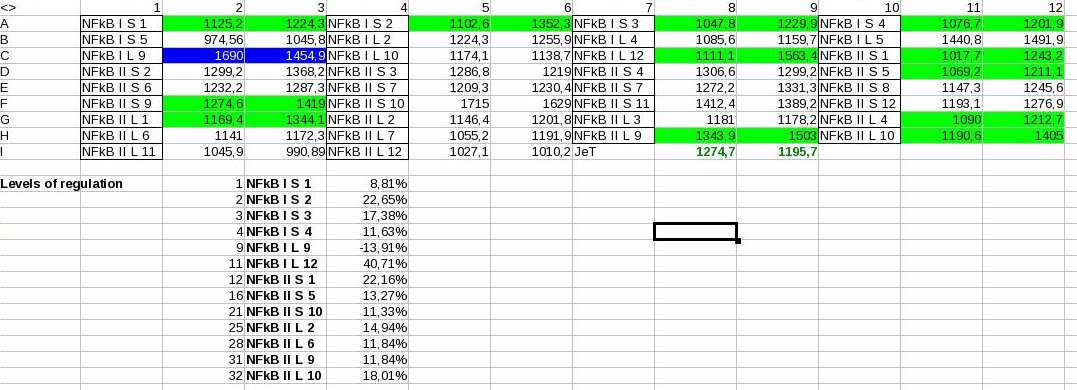
- Numerical evaluation of regulated (HIF, NFkB, p53) promoters: From 0 = no fluorescnence to 3 = strong fluorescence can be found in Results 9-1-2009
- HIF: Induction time was too short
- p53: Most cells were death, as the cells were left in camptothecine for too long
- Sequence analysis shows that the PCR protocol ("ROE-PCR") works (see images below), leaving the following remarks
- Long consesus motifs (e.g. p53) must be added in a higher proportion
- Obviously, small variations in pipetted oligo volume critically affects primer length annd composition (No other explanation for NFkB "promoters" being so short, compare Gel image and sequencing results).
This is the way forward
- Measure Const L 4, Const L 5, Const S 10, Const S 4 (+ Const L1, S5) → Four constitutive promoters of different strength and of our own making!)
- Transfect again:
- HIF S 3-20,23,24 ; HIF L 4,6-9,11,16,17,19,20,21,23 → Because bad choice of timepoint: Wait ~5h
- p53 S 8, 13, 22, 23, 24; p53 L 1, 3, 9, 11, 13, 17, 18, 19 → look promising, but bad choice of timepoint: Wait 30, 45, 60 minutes; alos induce by using UV light.
- 2nd round of promoter synjthesis
- Improving existing protocols
- p53 (higher content of p53 oligos)
- NfkB again (less start and stop?)
9-03-2009
New attempt to synthesize NFkB and p53 responsive promoters:
- Gene synthesis (compare Material and Methods) using the following oligo concentrations and a PCR purification step after the 1st seven cycles.
| p53
| NFkB I
| NFkB II
| Activator Mix
|
6µL p53 (O.91)
5µL p53 reverse (O.188)
1µL random (O.56)
1µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 3µL NFkB-1 (O.93)
3µL NFkB-2 (O.94)
5µL NFkB-rev (O.194)
1µL Random (O.56)
1µL Activator Mix
0.5µL Stop 5 new (O.187)
0.5µL Stop 3 (O.58)
| 3µL NFkB-1 (O.93)
3µL NFkB-2 (O.94)
4µL NFkB-rev (O.194)
3µL Random (O.56)
2µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 2µL each Ap1, Sp1, CREB (O.55, O.57, O.89)
1µL each NFY, Empty (O.90, O.95)
Water to 30µL
|
- Gel extraction. Excised two fragments per lane, one >1000 (L) and one <1000 and >150-200 (S)
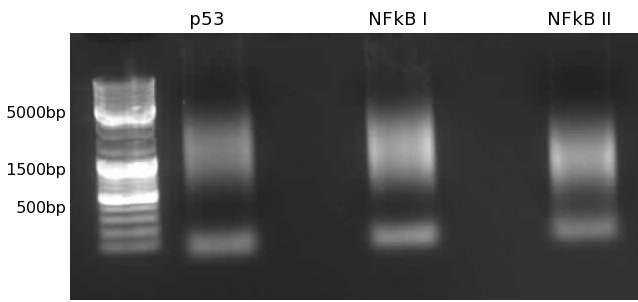 Synthesis of p53, NFkB (I and II) promoters1% agarose gel. Note that less short products exist due to lower concentrations of Stop 5 and Stop 3 oligos. 9-04-2009
New attempt to synthesize NFkB and p53 responsive promoters:
- Digest p31 with HinDIII, SpeI
- Digest yesterday's gel extractions (p53 S, p53 L, NFkB I S, NFkB I L, NFkB II S, NFkB II L) with HindIII, SpeI
- Ligation
- Transformation
Synthesis of HIF, p53 promoters from the first synthesis round:
- Screening of p53 again, 40 minutes after induction:
- Clone p53 L 17 and p63 L 19 show induction; they showed induction in the 1st screening also and will be transfected for FACS measurement
- Screening of HIF: HeLa Cell line not suited, dies too fast in minimal media -> to be repeated in MCF-7 cells
9-06-2009
New attempt to synthesize NFkB and p53 responsive promoters:
- Picked colonies for miniprepping:
- NFkB I S 6*
- NFkB I L 12*
- NFkB II S 12*
- NFkB II L 12*
- p53 S 12*
- p53 L 18*
9-07-2009
New attempt to synthesize NFkB and p53 responsive promoters:
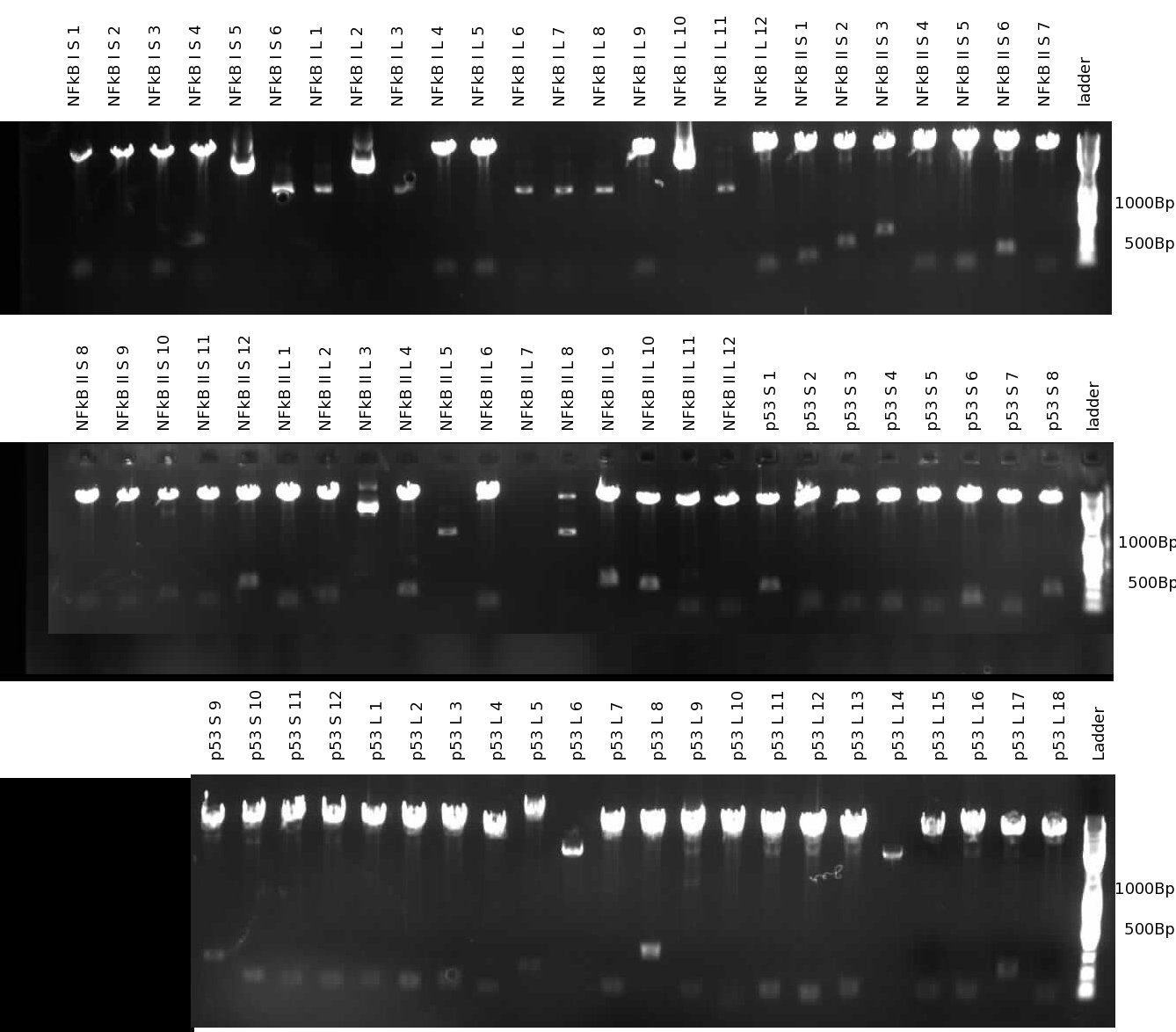 Synthesis of p53, NFkB (I and II) promoters - test digest1% agarose gel. Note that avg. product length is longer than in the previous synthesis round due to lower start/stop concentrations. 9-08-2009
New attempt to synthesize NFkB and p53 responsive promoters:
- Transfections (+/- TNF-a for NFkB, +/- xxx for p53)
9-09-2009
Synthesizing AHR, SREBP and pPARy
- Gene synthesis (compare Material and Methods) using the following oligo concentrations and a PCR purification step after the 1st seven cycles.
| SREBP
| AHR
| pPARγ
| Activator Mix
|
5µL SREBP (O.208)
4µL SREBP reverse (O.209)
1µL Sp1 (O.57)
2µL random (O.56)
1µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 5µL AHR (O.212)
4µL AHR reverse (O.213)
2µL random (O.56)
1,5µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 5µL pPARγ (O.210)
4µL pPARγ reverse (O.211)
2µL random (O.56)
1,5µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 2µL each Ap1, Sp1, CREB (O.55, O.57, O.89)
1µL each NFY, Empty (O.90, O.95)
Water to 30µL
|
- Gel extraction. Excised two fragments per lane, one >1000 (L) and one <1000 and >150-200 (S)
- Digest p.31, p48 with HinDIII, SpeI for 4h; 1h + SAP; Gel extraction
- Digest Gene synthesis products with HindIII, SpeI
- Ligation
Screening of NfKb, p53 responsive promoters (of second synthesis round)
- NFkB was transfected into U2OS and induced by addition of 2,5µM TNF-α
- p53 was transfected into MCF-7 and induced by addition of Camptothecin (CPT), a topoisomerase inhibitor
- Screening by eye / fluorescence microscopy
- NFkB: Promising clones. Screening will be repeated, including a second control (dye-free DMEM) as NFkB might be induced by starvation
- p53: Obviously, starvation did induce p53 whereas Camptothecin did not (maybe cells were harmed too much by the time p53 would have been active). Starvation does induce p53 as p53 is a target of AMP-Kinase. We will repeat the screening with a p53-inhibitor.
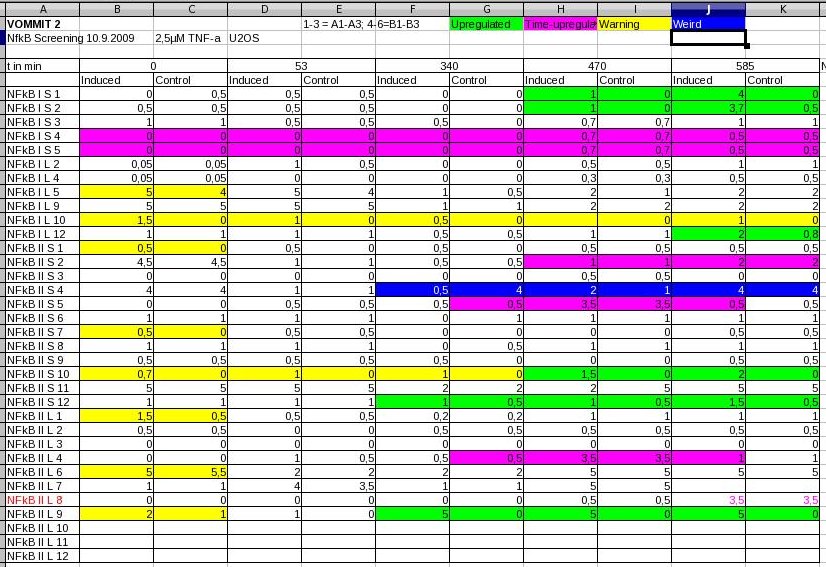 Screening of putative NFkB regulated promoters. 1-6 corresponds to relative GFP expression strength levels. 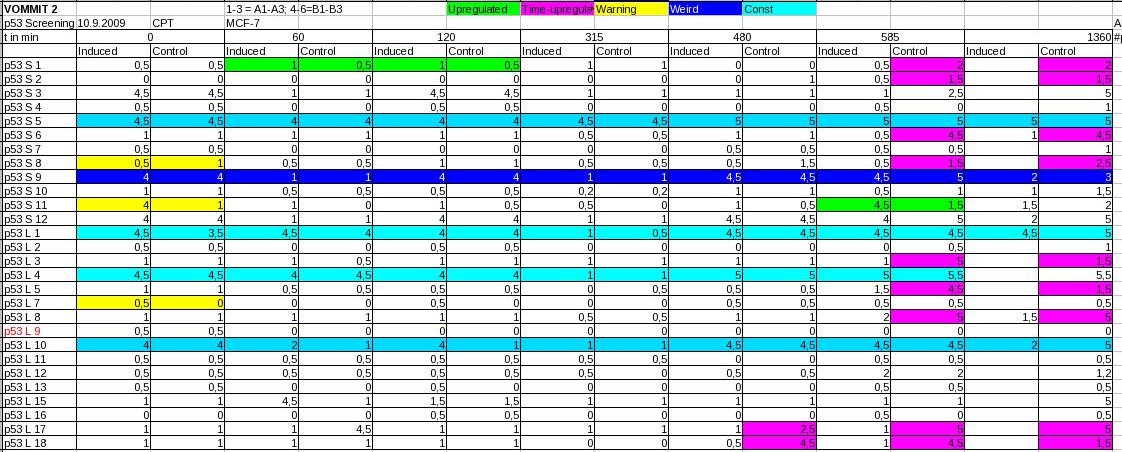 Screening of putative p53 regulated promoters. 1-6 corresponds to relative GFP expression strength levels. 9-10-09
9-14-09
- Minipreps
- Test digest to find only AHR synthesis is correct (pictures tbd)
9-15-09
- Induction of U2OS cells transfected with putative NFkB responsive promoters
- Screening by TECAN for NFkB of second synthesis round: Results show (a) that TECAN (despite initial advise by our favourite advisor telling us to screen by eye, instead of TECAN) and (b) many promoters show regulation. These will have to be measured in detail by flow cytometry. Also, for future TECAN screenings, a timepoint zero measurement will have to be added, but this series of measurement still is acceptable as comparable initial fluorescence levels were found by eye/microscopy as for previous screenings (data not shown).
 TECAN measurement results. A corning 96 well plate (transparent, flat) was used. Promoters upregulated by TNF-a are marked greeen, one promoter appears to be down-regulated (marked blue). - Repeat promoter synthesis for AHR, pPARγ, SREBP and Estrogen Receptor
9-16-09
- Ligation, Transformation of repeated nuclear receptor promoter synthesis
9-18-09
- Screening pf p53, HIF
- p53 screening showed: some promoters are downregulated under p53 induction, which is not how JeT (p31) behaves.
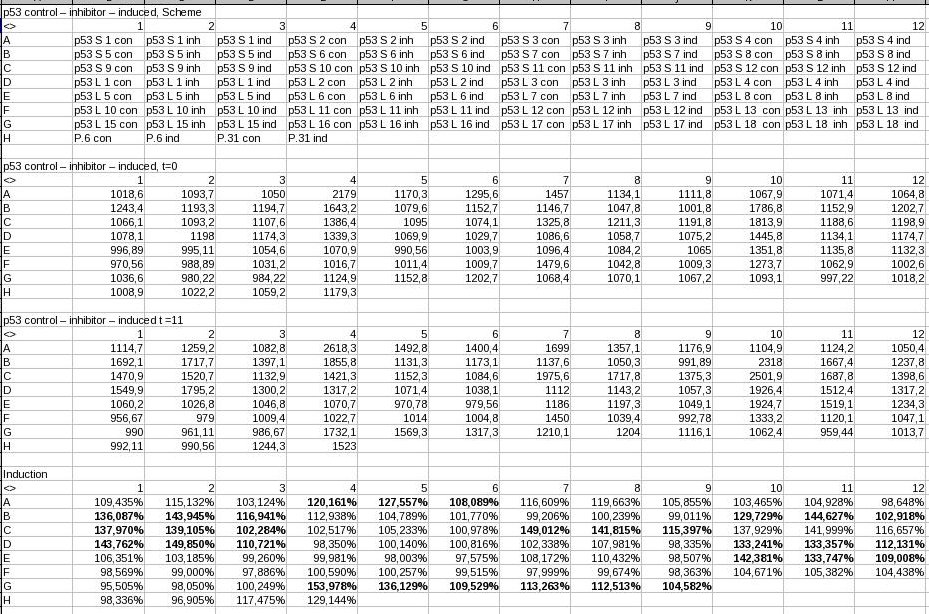 Screening of putative p53-regulated promoters by TECAN. the third table compares the values of the 1st with the values of the 2nd table. Promoters marked bold were selected for further characterization by flow cytometry. - p53 screening showed: some promoters are downregulated under p53 induction, which is not how JeT (p31) behaves.
 Screening of putative HIF-regulated promoters by TECAN. the third table compares the values of the 1st with the values of the 2nd table. Promoters marked bold were selected for further characterization by flow cytometry. 9-20-09
- Minipreps of second synthesis round for Estrogen Receptor, pPARγ and SREBP-regulated promoters
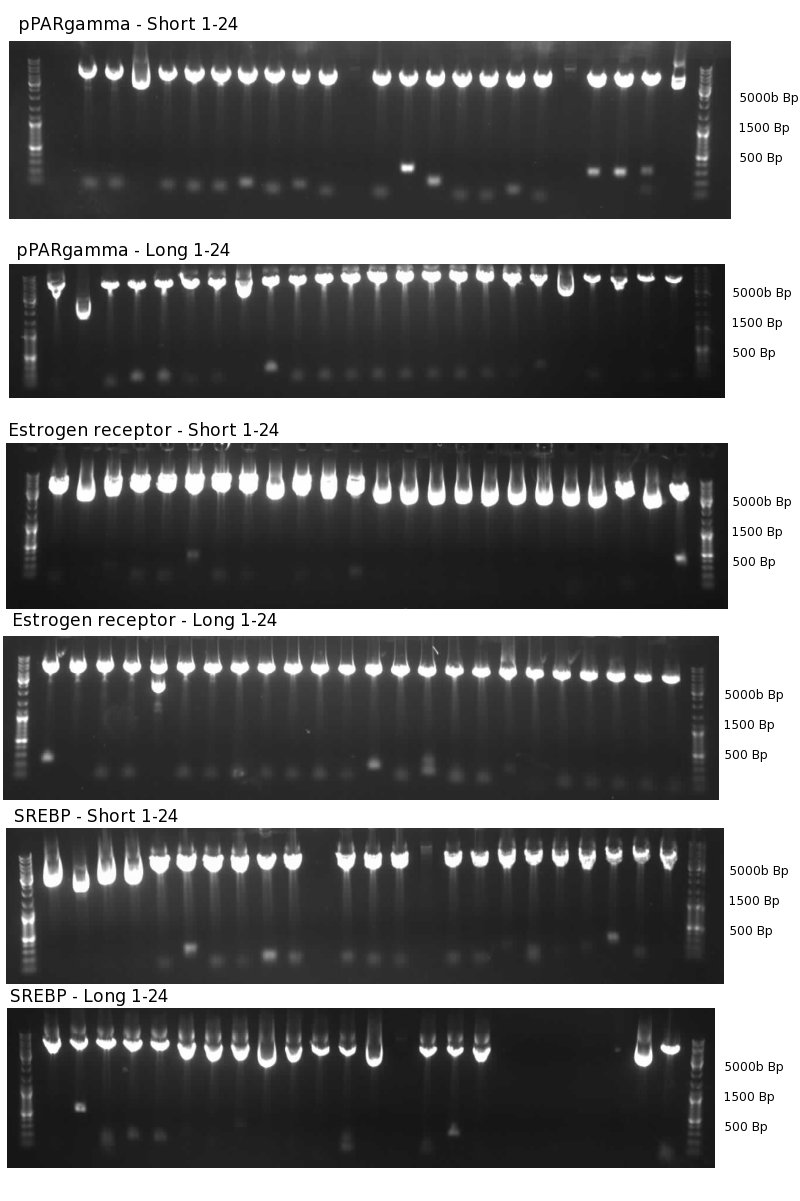 Test digest of second synthesis round for nuclear recptor regulated promoters 1% agarose gel. Digest with HindIII and SpeI, 1h 9-21-09
Synthesis was remade for the Promoters stated above. We improved gel extraction quality. Still, quality synthesis remained low. We selected clones from the gel that were subjected to screening subsequently. Later, we found out that probably damaged HinDIII enzyme was responsible for the bad synthesis quality.
9-30-09
- Screenings for AHR, Estrogen Receptor, pPARγ and SREBP-regulated promoters were finished in the previous days (compare Cell culture Notebook).
- For all indcution times, srugs and cell lines, compare Material and methods
- AHR results, first round of screening (with TCCD for inducion)
 putative AHR responsive promoters, screening by TECAN - AHR results, second round of screening (with Benzo[a]pyrene for induction)
 putative AHR responsive promoters, screening by TECAN  putative pPARγ responsive promoters, screening by TECAN - Estrogen Receptor results, first round of screening
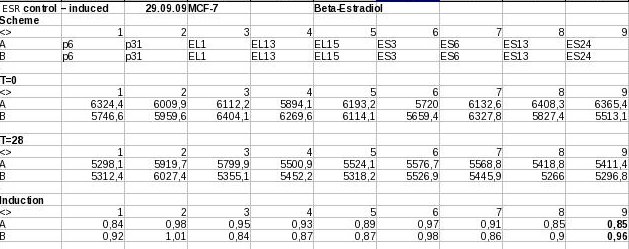 putative pPARγ responsive promoters, screening by TECAN
October 2009
In the weeks before wiki freeze, selected promoters were characterized by the measurement subgroup (see their Notebook ). At the same time, further screenings were conducted as described Also, some promoters were characterized roughly by triple TECAN-reads (namely, pPARγ and p53 responsive clones), or attempted to characterize in this was. We provide all spreadsheets thus generated in a two zip file: raw data and analyzed data. All Spreadsheets are commented with date of measurement. The only of those characterization which yielded reliable results we decided to publish on the registry is for pPARγ (see below or in analyzed data).
|
 "
"































