|
|
| Line 544: |
Line 544: |
| | <br> | | <br> |
| | <div class="heading"> | | <div class="heading"> |
| - | Descriptive Title of What You're Doing
| + | Some next steps on the modelling front |
| | </div> | | </div> |
| | <br> | | <br> |
| | <div class="desc"> | | <div class="desc"> |
| | </html> | | </html> |
| - | WIKI CODING HERE
| + | Today was dominated by meetings: first, Iman and I met with Anders to discuss where we're at on the project and what to do next. We ran our simulation and noticed that it shows that our GFP levels rise to infinity. This is because we haven't updated our model in a while and hadn't accounted for GFP degradation, but it also raises some interesting questions on the utility of our reporter circuit. If GFP:LVA doesn't degrade or takes a very long time to degrade, it may be very difficult to see any sort of response when we add AI-2 to the system. We also went over the sensitivity analysis and noticed that 2 of the parameters didn't really seem to be important in the overall rates of the system. More specifically, our system displayed a low degree of sensitivity to these parameters. What this means is that if we change the species involved in those reactions to something cheaper, or more readily available, or perform other tweaking, it shouldn't impede the overall performance of the system to any appreciable extent. The modelling team will meet again on Thursday morning to align our reactions and make sure that we really are modelling the same system. |
| | + | |
| | + | After the modelling meeting, we had a team Q and A session. This was very useful in (a) revealing what I don't know; and (b) clearing up these uncertainties. It also serves as excellent preparation for when we really have to perform on the Q and A front outside of our own team. |
| | + | |
| | + | I then worked more on our model, first by adding an element to account for GFP degradation and then approximating a degradation constant. For some reason, I had heard something to the effect of 20 seconds for a half-life with an LVA tag, and with that I was able to approximate a degradation constant. After all that, it occured to me to verify the half-life, and it turns out that I mixed it up with luciferase. GFP is much slower - around 40 minutes - which will be a problem if it is produced much more quickly than it degrades and if we surpass the saturation limits on our instrumentation. Tomorrow, I will approximate a degradation constant using the 40 minute value and work with the other members on the lab team to experimentally resolve the saturation limits on our machine. |
| | | | |
| | <html> | | <html> |
|
|
CAROL
Descriptive Title of What You’re Doing
WIKI CODING HERE
|
|
|
CHINMOYEE
Descriptive Title of What You're Doing
|
|
|
EMILY
Restriction Digest and Q & A Session
- Unfortunately my plates from yesterday of Vicki's construct in TOP10 (J13002-LuxOD47A-B0015) did not have any colonies. So I restrated the plasmid switch from the restriction digest, digesting both the construct in psB1AK3 and the psB1AC3 vector with XbaI and PstI restriction enzymes. I transformed the ligated product into TOP10 cells and plated on C plates. Hopefully these plates will have colonies tomorrow and I can proceed with a Colony PCR to verify that the construct is in the vector.
- Today we also had a team meeting where we had another Q & A session in preparation for aGEM and iGEM. I answered some general questions as well as some human practices questions. This was once again extremely helpful in identifying areas that I don't entirely understand, especially within some of the various sub-projects.
|
|
|
FAHD
Ethics & Outreach and Q & A
Today I continued to work on the ethics paper that I started yesterday. I also added extra information on the precautionary and the pro-actionary framework. I also made a program plan for the ethics conference that we will hold in Second Life ™. The plan includes information such as how we intend to carry out the conference, who are we going to invite, what type of question we will be asking our guest speakers, etc. .
We also had a Q & A with our team today. The questions were about our iGEM in general, synthetic biology, our project this year and specific questions concerning specific aspects of our project. The Q & A was an excellent exercise to prepare our team for the type of question that we will be asked during the aGEM and iGEM competition.
|
|
|
IMAN
Descriptive Title of What You're Doing
|
|
|
JAMIE
Descriptive Title of What You're Doing
|
|
|
JEREMY
Descriptive Title of What You're Doing
|
|
|
KATIE
Continuation of August 17th
I was able to move the DNA replication to the base of the spiral, but it seems I have lost permission on gyrase and cannot even delete the script inside so any changes will occur at a later date. I started the basic frame consisting of an RNA polymerase to make a complementary RNA strand and a single strand of DNA this polymerase can “read”. The difference in this animation is that specific bases must be used instead of generic nucleotide in order to make sense so the polymerase must know what the complementary nucleotide is.
I really want users to be able to pick what nucleotide is to be added to the mRNA strand so I believe I will work on a way to do this. For transcription and am in the process of making the polymerase move down the strand, but I may change how it runs at the moment.
I edited the second life blog update and sent it to receive feedback as well as a creative title before posting it and the pictures for it in the evening. I also continued with wiki updates for July and June, but now I believe I will have to make Thursday the day I finish the June updates while May has been completed to the best of my ability disregarding the week spent learning some molecular biology and lab techniques.
After the majority of team members went onto second life today, I was told that the prizes are given regardless of completing a lab mission. I do not have permission to view this script at this time, but I will request it. Also, the lab depends on notecards, which it seems we will need to properly test anything since the robots require giving a folder of objects including instructions that have yet to be added.
|
|
|
KEVIN
cPCR of Pqrr4+B0034+K082003
Yesterday, our reporter circuit with GFP:LVA ( Pqrr4+B0034+K082003) was constructed and transformed using colony 1 of Pqrr4+B0034 and already cut and freshly cut K082003. The K082003 part was already cut last time I attempted to construct this circuit, and this was used as well as a freshly cut K082003 to see if there are any differences between the two. On the plate of Pqrr4+B0034+K082003(old), there were noticeably less colonies than on the plate of Pqrr4+B0034+K082003(new). Perhaps most of the old K082003 linear DNA has degraded and was not cloned into Pqrr4+B0034, and most of the phosphotase treated vector remained as a linear piece of DNA, meaning the cells could not take up the plasmid that contains the antibiotic of interest. The following is the picture of the gel:
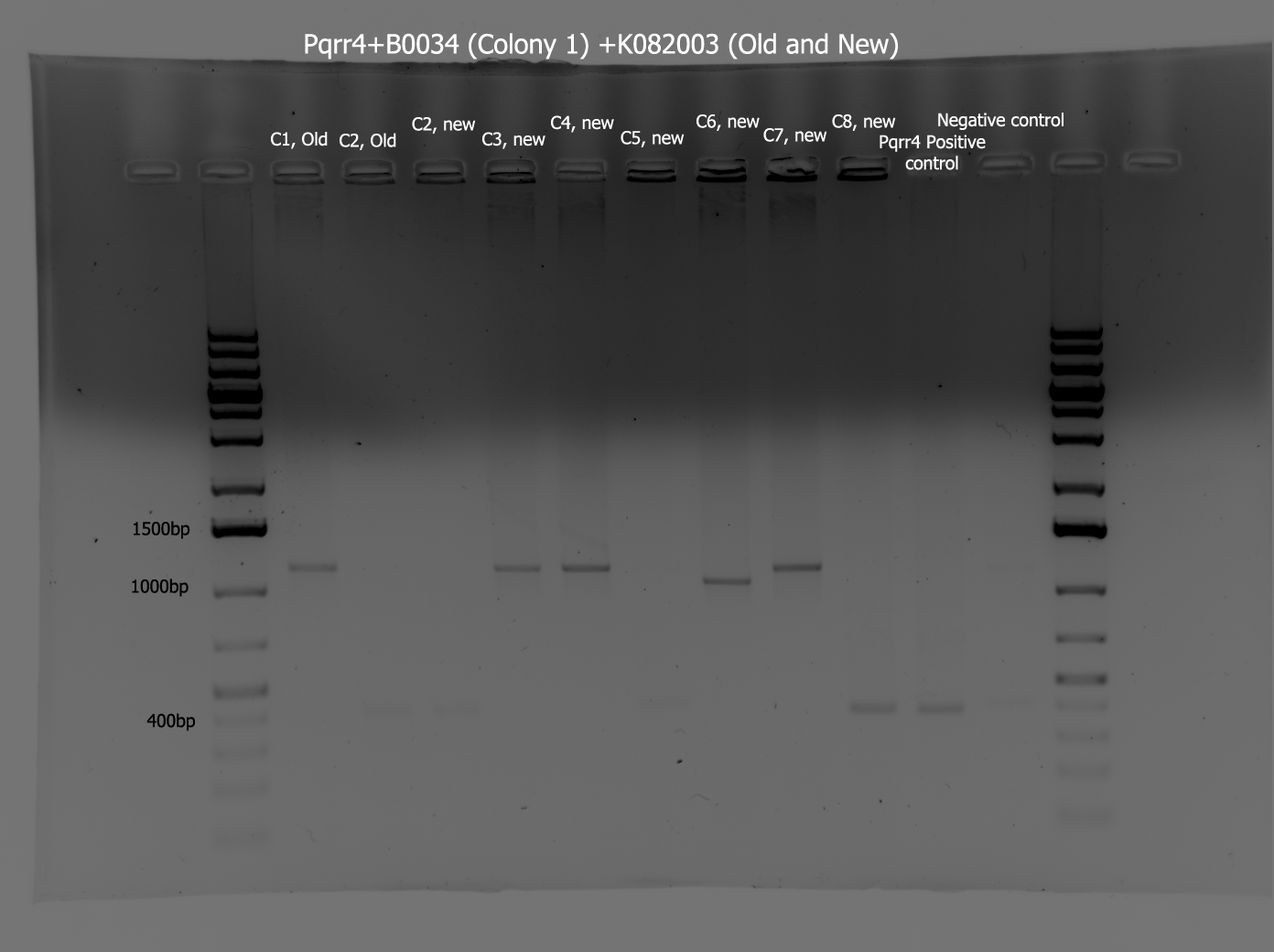
The band in (C1, old), (C3, new), (C4, new), and (C7, new) lanes seem to be at the expected size, at around 1170bp, including the additional bps that are due to the annealing of the Biobrick CP reverse primer. I will procede with these colonies tomorrow with Restriction digest to further verify the presence.
Measuring the fluorescence of the cells with the reporter and mutant circuits
Today, I played around with the plate reader. Blank LB broth with Kanamycin and Chloramphenicol antibiotics media was used to blank the machine. The following was the result:
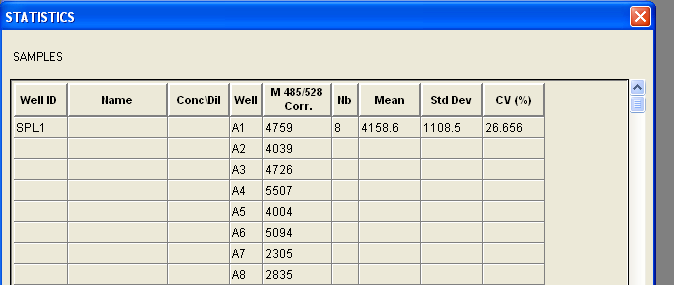
A1~A6 consists of the results from TOP10 cells with the reporter and mutant circuits. A7~A8 constists of the results from KT1144 cells with mutant circuits in them. According to http://www.mnstate.edu/provost/GFPPlateReaderAssayProtocol.pdf, the settings for the plate reader should be:
Excitation: 485/20 nm
Emission: 528/20
Just by looking at the results, it seems that the cells indeed do produce GFP. However, since I am yet to know what values I should be expecting to conclude that they are producing GFP, I cannot be sure; thus I will explore some papers in order to find some data to compare with.
|
|
|
MANDY
Descriptive Title of What You're Doing
|
|
|
PATRICK
Descriptive Title of What You're Doing
|
|
|
PRIMA
Descriptive Title of What You're Doing
|
|
|
STEFAN
Introducing the Team to Second Life
I finished up the Disease Hunting station in the Synthetic Kingdom. This was done by setting up 2 boxes which will rez the Bad Guy and Champion bacteria. This had to be set up in a way which will be controlled. This means putting the script to sleep for 3 seconds so people don't click on eat repeatedly and overload the sim with too many sensors (this may cause lag). I have also used a random rezzer so the interactions between the bacteria and also the cell will be more varied.
The whole team entered Second Life today which was very helpful. Having other people enter the Synthetic Kingdom made me realize any mistakes I may have made. For example, Jamie was confused about which bacteria to click. I then changed it to make it more clear that the bacteria all around are able to be clicked. In addition, some permissions weren't set and people couldn't move the vitamins and that was fixed. The GFP station still needs to be fixed but setting permissions on that is a little bit trickier.
|
|
|
VICKI
Some next steps on the modelling front
Today was dominated by meetings: first, Iman and I met with Anders to discuss where we're at on the project and what to do next. We ran our simulation and noticed that it shows that our GFP levels rise to infinity. This is because we haven't updated our model in a while and hadn't accounted for GFP degradation, but it also raises some interesting questions on the utility of our reporter circuit. If GFP:LVA doesn't degrade or takes a very long time to degrade, it may be very difficult to see any sort of response when we add AI-2 to the system. We also went over the sensitivity analysis and noticed that 2 of the parameters didn't really seem to be important in the overall rates of the system. More specifically, our system displayed a low degree of sensitivity to these parameters. What this means is that if we change the species involved in those reactions to something cheaper, or more readily available, or perform other tweaking, it shouldn't impede the overall performance of the system to any appreciable extent. The modelling team will meet again on Thursday morning to align our reactions and make sure that we really are modelling the same system.
After the modelling meeting, we had a team Q and A session. This was very useful in (a) revealing what I don't know; and (b) clearing up these uncertainties. It also serves as excellent preparation for when we really have to perform on the Q and A front outside of our own team.
I then worked more on our model, first by adding an element to account for GFP degradation and then approximating a degradation constant. For some reason, I had heard something to the effect of 20 seconds for a half-life with an LVA tag, and with that I was able to approximate a degradation constant. After all that, it occured to me to verify the half-life, and it turns out that I mixed it up with luciferase. GFP is much slower - around 40 minutes - which will be a problem if it is produced much more quickly than it degrades and if we surpass the saturation limits on our instrumentation. Tomorrow, I will approximate a degradation constant using the 40 minute value and work with the other members on the lab team to experimentally resolve the saturation limits on our machine.
|
 "
"












