SBML
[http://sbml.org The Systems Biology Markup Language (SBML)]is a computer-readable format for representing models of biological processes. It's applicable to simulations of metabolism, cell-signaling, and many other topics. SBML has been evolving since mid-2000 thanks to an international community of software developers and users. SBML does not represent an attempt to define a universal language for representing quantitative models. It would be impossible to achieve a one-size-fits-all universal language. A more realistic alternative is to acknowledge the diversity of approaches and methods being explored in systems biology, and seek a common intermediate format—a lingua franca—enabling communication of the most essential aspects of the models.
Why SBML?
The adoption of SBML offers many benefits, including:
- enabling the use of multiple tools without rewriting models for each tool.
- enabling models to be shared and published in a form other researchers can use even in a different software environment.
- ensuring the survival of models (and the intellectual effort put into them) beyond the lifetime of the software used to create them.
SBML is neutral with respect to programming languages and software encoding; however, it's oriented towards allowing models to be encoded using XML. By supporting SBML as a format for reading and writing models, different software tools (including programs for building and editing models, simulation programs, databases, and other systems) can directly communicate and store the same computable representation of those models. This removes an impediment to sharing results and permits other researchers to start with an unambiguous representation of the model, examine it carefully, propose precise corrections and extensions, and apply new techniques and approaches—in short, to do better science.
Software support
As the matter of fact, there has been lots of softwares known to us to provide some degree of support for reading, writing, or otherwise working with SBML. Thus, we make SBML to access our software, which makes it more compatible to other biological softwares. Users can output SBML file from our software, which means you can get a standard expression of biological system. It is facilitate for you to analyse the model in other SBML-related softwares.
To access the requirement, there must be a tool helps us to fetch the list of data from a SBML file. Fortunately, [http://sbml.org/Software/libSBML LibSBML] assists us for all the SBML related staff. LibSBML is an open-source programming library to help you read, write, manipulate, translate, and validate SBML files and data streams. The Latest stable release of LibSBML is 4.0.0. you can visit the SBML web site for more details. In terms of our software, the following three major functions has been accomplished based on LibSBML.
1.ReadSBMLFile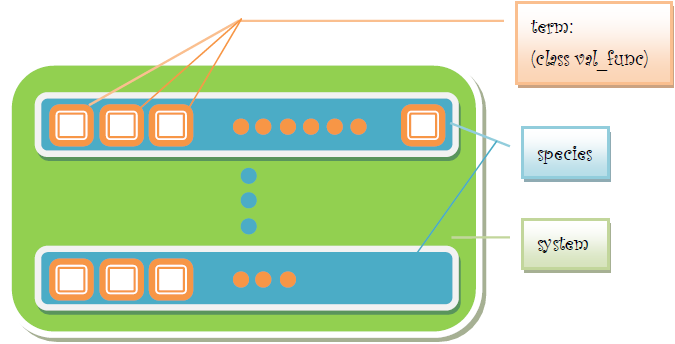 Schematic view of reading SBML The class ReadSBMLFile is used to extract all useful data from a SBML file and send them to the class “val_func”. Since each class represents one term of the the expression. all the terms will be push into a vector. In another word, another container has been created used to place a list of "val_func", of course you can realize that Nth list of "val_func" accommodates all the informations related to Nth species in this Biological system. Finally, all the containers will be packaged orderly and send to the Particle Swarm Optimization Algorithm (PSO) part or the Global Sensitivity Analysis (GSA) part for the next step. Here are the components of class “val_func”.
class val_func
{
public:
val_func();
~val_func();
int type; //the type of the kineticlaw
//Due to the ASTNode(a simple kind of data structure)
//used to store mathematical expressions in SBML
//most of them is algebraic expression), it is hard
//to judge the type mentioned in the file. Usually we
//give a number 15 to this variable, which implies the
//expression will be execute by route “user define”.
int spec_num; //number of species
int para_num; //number of parameters
vector<int> spec;//serial number of the species of the reaction
vector <double> para; //initial value of the parameters
vector <string> para_name; //IDs/names of the parameters
vector <string> var_name; //IDs/names of the species
map<int,double> min_data; //N/A
map<int , double> max_data; //N/A
const ASTNode * head; //ASTNode tree of the formula
double value( const vector<double>& s); //N/A
};
2.WriteSBMLFile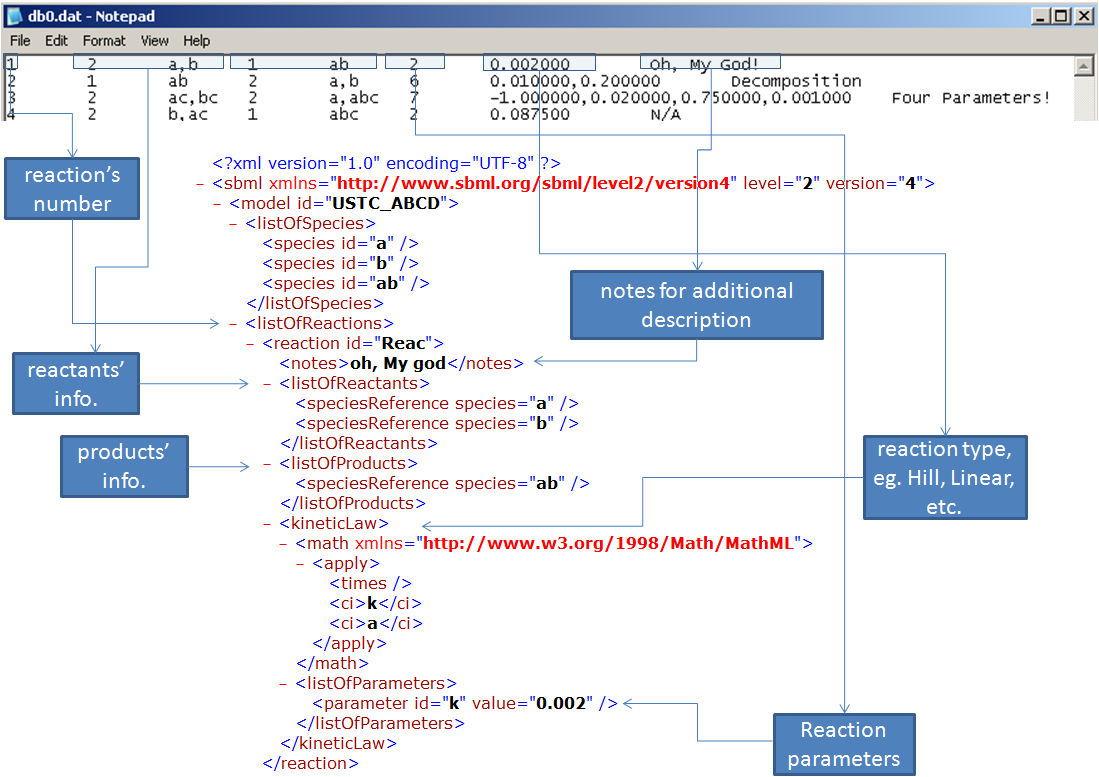 Convert from db0.dat to SBML file The class WriteSBMLFile is used to output a SBML file base on the species and parameters selected from the database. the amount of the exporting file is based on the users requirement.In this part, the class WriteSBMLFile extract data from the two following file: “subs.dat”; ”dbt.dat”.
Although we have listed all the types of bio-chemical reactions, it is also needed to make the mathematical expression fit each certain reaction. Since the SBML uses ASTNode(binary tree) to store the formulas, we choose to traverse binary tree and replace the name of each node the certain species and parameters. Thus, the kineticlaw is been made uniquely for each bio-chemical system.
Here are the steps of converting “subs.dat” and ”dbt.dat” to SBML file:
1. Create new SBMLDocument and model
2. Scrape the species names from "subs.dat", and create new species in model
3. Get reactions related information from"dbt.dat". As the graph, put them into the model by categories
4. Rename the node to species name corresponding
5. Validate the model
6. Output SBML file
3.verifying and other functions
One of the most important features of libSBML is its ability to perform SBML validation to ensure that a model adheres to the SBML specification for whatever Level+Version combination the model uses. Since it is difficult to ensure that everything in a model is constructed properly. The ability to perform automatic checks then becomes very useful.
LibSBML implements verification of SBML in two steps, represented by the two methods SBMLDocument::checkInternalConsistency() and SBMLDocument::checkConsistency(). The former verifies the basic internal consistency and syntax of an SBML document, and the latter implements more elaborate validation rules (both those defined by the SBML specifications, as well as additional rules offered by libSBML). When an application builds up a model programmatically and is finally ready to say "yes, this model is finished", it should call both of these methods to help ensure the correctness and consistency of the finished result. Conversely, if it is reading a model from a file or data stream, the function will automatically verify the syntax and schema consistency of the document. The application only needs to check the results by interrogating the error log on the SBMLDocument object, then call SBMLDocument::checkConsistency() for checking the model against the SBML validation rules.
Assitant Input Plug-ins
In order to simplify the input procedure of the data file describing time course which sometimes gets really redundant, we also program a small .exe file to help convert *.bmp file to *.dat file based on following assumptions.
- The x-axis and y-axis are already unified to the range [0,1], while any other range could be realized by multiple additional factors on the unified range.
- The program will only identify black pixels on the picture, and for any given x value, the y value is decided by the average of all existing points corresponding to that same x value.
- Color depth sould be no less than 8 bits.
- Sampling step length is determined by 1 divided by the horizontal pixel numbers.
Following is a small demo of this tool.
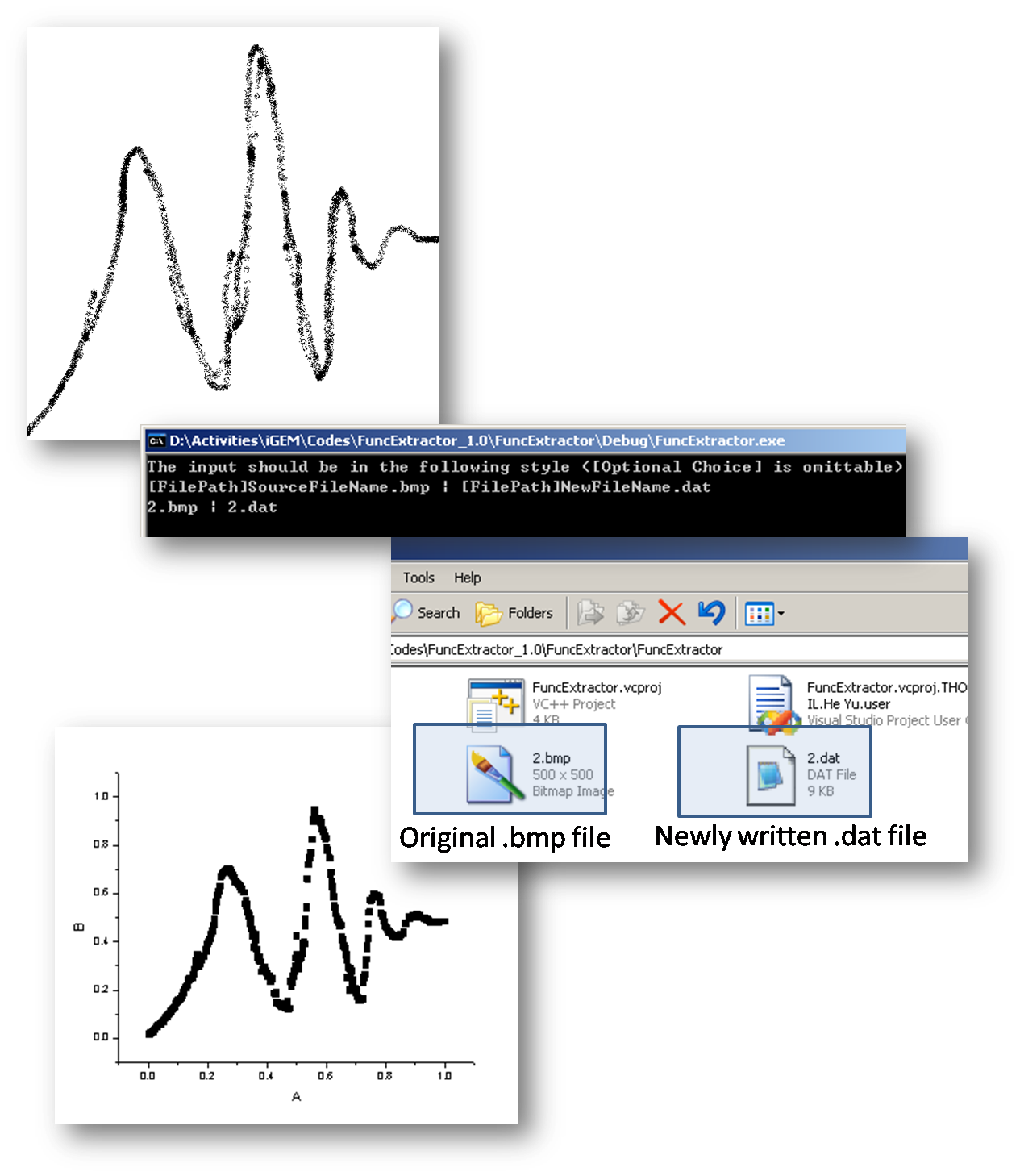 Arbitrarily draw a curve in painting tools under windows xp There are definitely quite a lot to improve, including
1.Rescale-able range of x-axis and y-axis
2.User designated step length
3.Function of ‘smoothing’
4.Identify the curve based on contrast rather than requiring the effective pixel to be black.
|  "
"
























