Material
Kits
| Kit | supplier
|
| |
|
| CompactPrep Plasmid Maxi Kit | Qiagen
|
| High Pure PCR Template Preparation Kit | Roche
|
| HISpeed Plasmid Maxi Kit | Qiagen
|
| QIAprep Spin Mini Kit | Qiagen
|
| QIAquick Gel Extraction Kit | Qiagen
|
| QIAquick PCR Purification Kit | Qiagen
|
| RNeasy Plant Mini Kit | Qiagen
|
| QuantiFast Probe RT-PCR Kit | Qiagen
|
| SensiMix One-Step Kit | Quantace
|
| QuikChange XL Site-Directed Mutagenesis Kit | Strategene
|
Marker
| Marker | supplier
|
| GeneRuler™ High Range DNA Ladder | MBI Fermentas
|
| GeneRuler™ 1kb DNA Ladder Mix | MBI Fermentas
|
Enzymes
| Enzym | supplier
|
| Pfu DNA polymerase | Stratagene
|
| Phusion DNA polymerase, Mastermix 2x | Finnzymes
|
| Taq DNA polymerase | MBI Fermentas
|
| T4 DNA ligase | MBI Fermentas / New England Biolabs
|
| Reverse transcriptase | Quantace
|
Restriction Enzymes
| Enzym | supplier
|
| BclI | MBI Fermentas
|
| EcoRI | New England Biolabs
|
| HindIII | New England Biolabs
|
| MfeI | New England Biolabs
|
| NheI | New England Biolabs
|
| NotI | New England Biolabs
|
| PstI | New England Biolabs
|
| ScaI | New England Biolabs
|
| SpeI | New England Biolabs
|
| SspI | New England Biolabs
|
| SphI | New England Biolabs
|
| XbaI | New England Biolabs
|
Bacteria
| E.coli strain | usage | reference
|
| DH5a | amplification of plasmids | Invitrogen
|
| SCS110 | amplification of unmethylated plasmids | Stratagene
|
Bacteria Growth Media
Luria Broth (LB)
- 10 g/L tryptone
- 5 g/L yeast extract
- 10 g/L NaCl
| Antibiotic | concentration
|
| Ampicillin | 100 mg/ml
|
| Kanamycin | 50 mg/ml
|
Eucaryotic cell lines
Cell Culture Media
DMEM+++
- DMEM
- 1% Glucosamin
- 1% Pen/Strep
- 10% FCS
DMEM++
- DMEM
- 1% Glucosamin
- 1% Pen/Strep
- 1% non-essential aminoacids
Zeocin-Medium
Hygromycin-Medium
- DMEM+++
- 200 µl/ml Hygromycin
Minimal Medium
- 2,1 g/l NaHCO3
- 1µM CaCl2
- 10 µM Hepes-Buffer [1]
- 1 % Pen/Strep
- Krebs-Henseleit-Buffer (Sigma-Aldrich [http://www.sigmaaldrich.com/etc/medialib/docs/Sigma/Product_Information_Sheet/1/k3753pis.Par.0001.File.tmp/k3753pis.pdf])
Oligos
We used [http://www.lablife.org LabLife], a great, non-commercial lab management resource for lab management and especially for keeping track of Oligos we used. All Oligos therefore are numbered, and all Notebook entries describing PCR reactions refer to a certain number of Oligo. A complete list of all Oligos we had in our lab can be downloaded as comma-separated values; find below a list of more interesting oligos we used for RA-PCR and LAM-PCR.
Oligos used for RA-PCR
| Name
| Sequence
|
| Stop 5'
| CAGTACTAGTGGGTGACGGGTTCA
|
| Stop 3'
| TGACAAGCTTAGTGAACCCGTCACCC
|
| Sp1 responsive (general transactivator)
| GCGATCGGCAGATCAGGGGCGGGGCGGGTGACGGGTTCA
|
| Ap1 responsive (general transactivator)
| GCGATCGGCAGATCATGACTCAGGGTGACGGGTTCA
|
| CREB responsive (general transactivator)
| GCGATCGGCAGATCABNBVNTGACGTCAGGGTGACGGGTTCA
|
| NFY responsive (general transactivator)
| GCGATCGGCAGATCANNCCAATNNGGGTGACGGGTTCA
|
| Spacer forward
| GCGATCGGCAGATCANNNNNNNNNNGGGTGACGGGTTCA
|
| Spacer reverse
| TGATCTGCCGATCGCNNNNNNNNNNTGAACCCGTCACCC
|
| AHR responsive, forwards
| GCGATCGGCAGATCACCYCNRRSTNGCGTGASAGGGTGACGGGTTCA
|
| AHR responsive, reverse
| TGATCTGCCGATCGCASAGTGCGNTSRRNCYCCTGAACCCGTCACCC
|
| Estrogen Receptor responsive, forward v 1
| GCGATCGGCAGATCAGGTCACTGTGACCGGGTGACGGGTTCA
|
| Estrogen Receptor responsive, forward v 2
| GCGATCGGCAGATCARGNCANNNTGACCYGGGTGACGGGTTCA
|
| Estrogen Receptor responsive, reverse
| TGATCTGCCGATCGCCCAGTGTCACTGGTGAACCCGTCACCC
|
| HIF responsive, forward v 1
| GCGATCGGCAGATCATCTGTACGTGACCACAGGGTGACGGGTTCA
|
| HIF responsive, forward v 2
| GCGATCGGCAGATCAGTCTACGTGCGGACGGGTGACGGGTTCA
|
| NFκB responsive, forward v 1
| GCGATCGGCAGATCAGGGGACTTTGCCGGGTGACGGGTTCA
|
| NFκB responsive, forward v 2
| GCGATCGGCAGATCAGGGGRWYYCCCGGGTGACGGGTTCA
|
| NFκB responsive, reverse
| TGATCTGCCGATCGCCCGTTTCAGGGGTGAACCCGTCACCC
|
| p53 responsive, forward
| GCGATCGGCAGATCAGAACATGTCCAAGCATGCTGGGGTGACGGGTTCA
|
| PPARγ responsive, reverse
| TGATCTGCCGATCGCHACTGGAAACTGGRRTGAACCCGTCACCC
|
| PPARγ responsive, forward
| GCGATCGGCAGATCARRGGTCAAAGGTCAHGGGTGACGGGTTCA
|
| SREBP responsive, forward
| GCGATCGGCAGATCAKATCACCCCACGGGTGACGGGTTCA
|
| SREBP responsive, reverse
| TGATCTGCCGATCGCCACCCCACTAKTGAACCCGTCACCC
|
| p53 reverse
| TGATCTGCCGATCGCGTCGTACGAACCTGTACAAGTGAACCCGTCACCC
|
| HIF reverse
| TGATCTGCCGATCGCGATCCGTCGTGCATCAGTGTGAACCCGTCACC
|
Oligos for LAM-PCR
| Name
| Sequence
|
| LAMPCR9_1 (biotinylated)
| AATACCGCGCCACATAGCAG
|
| LAMPCR9_2 (biotinylated)
| TTCTTCGGGGCGAAAACTCT
|
| LAMPCR9_3 (biotinylated)
| CACTCGTGCACCCAACTGAT
|
| LAMPCR9_4
| TCTGGGTGAGCAAAAACAGGA
|
Oligos for real-time RT-PCR
| Name
| Sequence
|
| GAPDH_forward_primer
| GGTATCGTGGAAGGACTCATGACC
|
| GAPDH_reverse_primer
| GATGACCTTGCCCACAGCCTTG
|
| GAPDH_probe
| CTCTCCAGAACATCATCCCTGCCTCTAC
|
| G6PD_forward_primer
| GAGTCCTGCATGAGCCAGATAGG
|
| G6PD_reverse_primer
| GCACCATGAGGTTCTGCACCATC
|
| G6PD_probe
| ACCAGATCTACCGCATCGACCACTACCT
|
| 18s_rRNA_forward_primer
| GGCCCTGTAATTGGAATGAGTCCAC
|
| 18s_rRNA_reverse_primer
| GCTCCCAAGATCCAACTACGAGCTT
|
| 18s_rRNA_probe
| CAGCAGCCGCGGTAATTCCAGCTCC
|
| G2TUB_forward_primer
| TTCTACCAGGCAGACGATGAGCAC
|
| G2TUB_reverse_primer
| CCCAGTTGTTGCCAGCTCCTC
|
| G2TUB_probe
| CTGCTGGACTTGGAACCCCGGGTG
|
| bACT_forward_primer
| CAGGCACCAGGGCGTGATGG
|
| bACT_reverse_primer
| CTCCATGTCGTCCCAGTTGGTG
|
| bACT_probe
| AGGATTCCTATGTGGGCGACGAGGC
|
| eGFP_forward_primer
| CGACCACATGAAGCAGCACGAC
|
| eGFP_reverse_primer
| CGATGCCCTTCAGCTCGATGC
|
| eGFP_probe
| AGGCTACGTCCAGGAGCGCACCATC
|
| mCherry_forward_primer
| CAAGTGGGAGCGCGTGATGAAC
|
| mCherry_reverse_primer
| CCAGCCCATGGTCTTCTTCTGC
|
| mCherry_probe
| TGACCGTGACCCAGGACTCCTCCC
|
Plasmids
The following is a list of all used plasmids. Some of them are just working plasmids, others are of greater value for our project. The latter are bold in the table.
| Plasmid Abbreviation | plasmid name | insert | remarks | resistance
|
| P1 | pcDNA5/FRT | | | Ampicillin/Hygromycin
|
| P2 | pEGFP template | | | Ampicillin
|
| P3 | mcherry template | | | Kanamycin
|
| P4 | mcherry ΔPstI | | | Kanamycin (?)
|
| P5 | pcDNA5/FRT ΔPstI | | | Ampicillin/Hygromycin
|
| P6 | pcDNA5/FRT ΔPstI/ΔEcoRI | | negative control for measurements | Ampicillin/Hygromycin
|
| P7 | pcDNA5/FRT ΔPstI/ΔEcoRI *unmethylated* | - | transformed in SC110 | Ampicillin/Hygromycin
|
| P8 | Part:BBa_E0040 | E0040 (GFPmut3B) | does not seem to work in eukaryotes | Ampicillin
|
| P9 | pFRT/lacZeo | - | plasmid was stably integrated in cell lines and used for stable integration of constructs | Ampicillin/Zeocin
|
| P10 | pcDNA5/FRT ΔPstI/ΔEcoRI EGFP | EGFP (from pEGFP-N1) | | Ampicillin/Hygromycin
|
| P11 | pcDNA5/FRT ΔPstI/ΔEcoRI mcherry JeT | mCherry, Jet promoter | | Ampicillin/Hygromycin
|
| P12 | pcDNA5/FRT ΔPstI/ΔEcoRI mcherry Min | mCherry, Min promoter | | Ampicillin/Hygromycin
|
| P13 | pcDNA5/FRT ΔPstI/ΔEcoRI GFPb3mut | E0040 (GFPmut3B) | | Ampicillin/Hygromycin
|
| P23 | psB1A3 - Registry | J04450 (RFP device) | backbone used for creation of BBb submission plasmid | Ampicillin
|
| P27 | pEGFP-N1 | EGFP | preliminary (+) control for transfection | Kanamycin
|
| P29 | pSB1A3 - Registry | P1010 | | Ampicillin
|
| P30 | pcDNA5/FRT ΔPstI/ΔEcoRI GFP Min | GFP, Min promoter | | Ampicillin/Hygromycin
|
| P31 | pcDNA5/FRT ΔPstI/ΔEcoRI GFP BBbJeT | GFP, standardized JeT promoter | | Ampicillin/Hygromycin
|
| P32 | CFP_emp_cytosole | CFP, cytosole localization signal | template | Kanamycin
|
| P33 | YFP_emp_cytosole | YFP, cytosole localization signal | template | Kanamycin
|
| P34 | YFP_GPI_PM | YFP, PM localization signal | template | Kanamycin/Neomycine
|
| P35 | CFP_GPI_PM | CFP, PM localization signal | template | Kanamycin/Neomycine
|
| P36 | YFP_Sar1_ER | YFP, ER localization signal | template | Kanamycin
|
| P37 | CFP_Sar1_ER | CFP, ER localization signal | template | Kanamycin
|
| P38 | EBFP_NLS_nucleus | EBFP, nuclear localization signal | template | Kanamycin (?)
|
| P39 | eYFP_NLS-cas8_nucleus | EYFP, nuclear localization signal | template | Kanamycin (?)
|
| P40 | mCherry_H2B_nucleus | mCherry, nuclear localization signal | template | Kanamycin (?)
|
| P41 | eYFP_H2B_nucleus | EYFP, nuclear localization signal | template | Kanamycin (?)
|
| P42 | GFP_myrpalm_PM | GFP, PM localization signal | template | Kanamycin (?)
|
| P43 | mCherry_myrpalm_PM | mCherry, PM localization signal | template | Kanamycin (?)
|
| P44 | GFP_mitomeet_Mitochondria | GFP, mitochondrial localization signal | template | Kanamycin (?)
|
| P45 | pOG44 | FLP | contains FLP enzymes that is used for stable integration of pcDNA5/FRT derived plasmids at FRT sites in the genome | Ampicillin
|
| P46 | Oakes NFkB | 5x NFκB responsive element (?) | backbone: pGL3 | Ampicillin
|
| P47 | Oakes NFAT | NFAT RE | backbone: pGL4 | Ampicillin
|
| P48 | p31_cmv_core_Jet_prox | CMV core promoter, JeT proximal promoter | standardized (BBb) fused promoter | Ampicillin/Hygromycin
|
| P49 | BBb_JeT pSB1A3 | standardized JeT promoter | backbone: pSB1A3; our very first plasmid to be send to the registry! | Ampicillin
|
| P51 | pcDNA5/FRT ΔPstI/ΔEcoRI BBbJeT | standardized JeT promoter | | Ampicillin/Hygromycin
|
| P52 | hsp70-EGFP | EGFP, hsp70 promoter (?) | natural heat sensitive promoter | (?)
|
| P53 | pcDNA/FRT ΔPstI/ΔEcoRI BBbCMV | standardized CMV promoter | | Ampicillin/Hygromycin
|
| P55 | pcDNA5/FRT ΔPstI/ΔEcoRI GFP_BBbCMV | GFP with standardized CMV promoter | exactly the same plasmid as P31 except for the promoter | Ampicillin/Hygromycin
|
| P56 | p31 CYP1A1 | GFP, CYP1A1 promoter | natural promoter | Ampicillin/Hygromycin
|
| P57 | pcDNA/FRT ΔPstI/ΔEcoRI mCherry_BBbJeT | mCherry with standardized JeT promoter | same plasmid as P31 except for fluorescent protein; used for cotransfection for measurements | Ampicillin/Hygromycin
|
| P58 | intron in submission plasmid | | | Ampicillin
|
Instruments
The following is a list of all used Instruments.
| Instrument | Name | Manufactor
|
| Table Centrifuge | Microfuge® 18 Centrifuge | Beckman CoulterTM
|
| Table Centrifuge | MicrofugeR 22® Centrifuge | Beckman CoulterTM
|
| Centrifuge | Allegra X-12 | Beckman CoulterTM
|
| Shaker | VORTEXGENIER 2 | Scientific Industries, SITM
|
| Heating plate (magnet stirrer) | MR Hei-Standard | Heidolph
|
| Heatblock | QBD4 | Grant
|
| Heatblock (shakeing function) | Thermomixer comfort | Eppendorf
|
| PCR-machine | MyCyclerTM thermo cycler | BioRad
|
| UV-chamber | Transluminator | Vilber Lourmat
|
| Scale (fine) | PioneerTM PA114C | OHAUS
|
| Scale | PioneerTM PA4101C | OHAUS
|
| Fridge | KTP 1750 Premium | Liebherr
|
| Freezer | GP 1366 Premium | Liebherr
|
| -86 Freezer | -86°C Ultralow Freezer | NUAIRETM
|
| Hood | Tischabzug | Wesemann® Laboreinrichtung
|
| Draw-off pump | Vacuhand control | Vacubrand
|
| Incubator | HT Multitron Version 2 | INFORS
|
| Incubator (cell culture) | HERAcell 150 | Thermo electron coorperation
|
| Hood (cell culture) | HERAsafe | Thermo electron coorperation
|
| Plate reader | Infinite M200 | TECAN
|
| FACS | Cytomics FC 500 MPL | Beckman CoulterTM
|
| Microscope | CKX41 | Olympus
|
| Computer | | Sun Microsystems
|
| UV/Visivle Spectrometer | Ultrospec 3300 pro | Amersham Biosciences
|
| Photometer | NanoDrop® ND-1000 Spectrophotometer | peQLab Biotechnologie GmbH
|
| Photometer | NanoVueTM | General Electric
|
| Ice Machine | MF22 | SCOTSMAN®
|
| Gel electrophoresis chamber | Mupid®-One | Advance
|
| pipetting robot | QIAcubeTM | QIAGEN
|
| Wide field microscope | Nikon Eclipse 90i | Nikon
|
| PCR machine | MyCycler thermal cycler | BIO-RAD
|
| Real-time PCR machine | StepOnePlusTM Real-time PCR System | Applied BiosystemsTM
|
Lab material
The following is a list of the used Lab material.
| Material | Name | Manufactor
|
| Bottle 50 ml, 100 ml, 200 ml, 500 ml, 1000 ml | Schott Duran | Schott
|
| Beaker 50 ml, 100 ml, 200 ml, 500 ml 1000 ml | - | Simax
|
| Conical flask 500 ml, 1000 ml, 100 ml | FB33141, FB33142, FB33139 | Fischerbrand
|
| Conical flask 300 ml | - | Simax
|
| Single channel pipet | Pipetman® P2, P20, P200, P1000 | Gilson
|
| Multichannel pipet | Research® Multikanal | Eppendorf
|
| Multistep pipet | e10, e120, e1000, e5000 | Biohit
|
| Pipet | accu-jet® pro | Brand
|
| PCR-tubes | PCR softtubes | Biozym
|
| Microcentrifuge tubes | safe-lock-tubes 2 ml/1,5 ml | Eppendorf
|
| Centrifugetubes | BD FalconTM 15 ml /50 ml | BD Biosciences
|
| PCR 8-strips | Tube Strips, CLR | Biorad
|
| Serological Pipet | 5 ml, 10 ml, 25 ml | BD Labware
|
| Gloves | Latex Medical Examination Gloves (S,M,L) | Blossom
|
| Nitrile Gloves | FreeformTM SE Examination Gloves (S,M,L) | MICROFLEX®
|
| Filtertips | Extended length Filtertips | Starlab
|
| Pipettips | TipOne® | Starlab
|
| Counting Chamber | Neubauer | Brand
|
| Inoculating loop | - | nuncTM
|
| Disposal bags | - | Plastibrand®
|
| Flasks | Tissue Culture Flasks | TPP®
|
| Plate | Microplates for life science Research | PerkinElmerTM
|
| IBD | Lab-Tek® | nuncTM
|
| 6/16-Well plates | NunclonTM Surface | nuncTM
|
Methods
General Methods
Transformation of Bacteria
For enrichment of vectors, E. coli DH5α were used. For the transformation 100 µl of the competent cells were thawed on ice and 50 – 400 ng DNA solution added (depending on the concentration of the DNA solution). After a 30-60 minute incubation on ice, cells were made permeable for the DNA by heat shocking for 45 seconds at 42 °C and a further 3 minute incubation on ice. The samples were than rescued by adding 500 µl preheated antibiotic free LB-medium and incubated for one hour at 37 °C while shaking for induction of the antibiotic resistance. The selection for plasmid containing and therefore antibiotic resistant bacteria was conducted by plating them on antibiotic containing LB-agar plates.
Glycerol stock
To store bacteria for long term glycerol stocks were used. Therefore 1 ml of an over night culture were added to 150 µl of 80 % Glycerol into a cryo tube, vortexed and incubated at room temperature for 30 min. Afterwards the glycerole stock was stored at -80 °C.
Isolation of plasmid DNA by alkaline lysis (mini and maxiprep)
For analysis of ligations and transformations QIAprep Spin Kits (Qiagen, Hilden) were used following the manufacturer instructions. For miniprep a single colony was picked from a LB-agar plate or glycerol stock was used to inoculate 5 ml LB-medium with appropriate antibiotic for selection (100 µg/µl ampicillin, 50 µg/µl kanamycin, 35 µg/µl chloramphenicol). Bacteria were grown over night at 37 °C while shaking (200 rpm). By using 4 ml over night culture with this kit the yield was around 6-10 µg. For maxipreps the Qiagen CompactPrep Plasmid Maxi Kit was used following the instructions given by the instruction manual. In this case 250 ml LB-medium were inoculated and used for preparation of plasmid DNA. The routinely yield was 300-400 µg plasmid DNA. Purity and amount of DNA was analysed using a NanoDrop.
Preparing chemically competent cells
First, a 20 ml over night culture was inoculated in antibiotic free LB medium from a fresh single colony and transferred into 400 ml antibiotic free LB medium the next day. This culture was incubated at 37 °C while shacking until an OD600 of 0.5 – 0.6 was achieved. The culture was than cooled down on ice, centrifuged (8 min, 4 °C, 3500 rpm), the supernatant discarded and the pellet resuspended in 10 ml 100 mM CaCl2. After addition of further 190 ml 100 mM CaCl2 the suspension was incubated on ice for 30 min. The suspension was than again centrifuged (8 min, 4 °C, 3500 rpm), the supernatant discarded, the pellet resuspended in 20 ml 82.5 mM CaCl2 with 17.5 % glycerol and aliquoted. The aliquots were flash frozen in liquid nitrogen and than stored at -80 °C until usage.
Ligation
Standard Ligation Protocol
T4 ligase joins the 5' phosphate and the 3'-hydroxyl groups of DOUBLE stranded DNA molecules. First step for ligation is to estimate the vector and insert concentrations:if insert is from PCR, assume that 50% is recovered, typically 5 mg, and resuspended in 10 ml of H20; same with vector, assume half is recovered in purification/precipitation and resuspend in 10-20 ml of H20 if it is not re-suspended already. Second step is to plan control reactions: One reaction with no insert, one reaction with no vector (if enough insert is available). Then Molar Ratio of Insert to Vector is determined where a ratio of Insert:Vector 3:1 (100-150ng Vector DNA) is tried to be achieved. After this, ligation mixure is setup in the following way:
- 10X Ligase buffer (Promega #M1801)
- 1 ml T4 Ligase (Promega #M1801, 3 U/ml)
- Bring volume to 10 (or 20 see note below) ml with nuclease-free water.
- If doing a blunt-end ligation, it may be needed to add PEG (up to 15%) to increase the efficiency.
Samples are incubated according to the guidelines below
- Incubate sticky end ligation reactions at room temperature for 3 hours (or at 4 to 8 °C, overnight).
- Incubate blunt-end ligation reactions at 17 °C for 4 to 18 hours.
After incubation the ligase is heat inactivated by placing tube in 65 °C water bath for 10 minutes. 2 ml of the ligation mixture is used for transformation.
Transformed cells are plated on selective media and incubated overnight.
Site-directed mutagenesis
For removal of unwanted Restriction Sites, a PCR-based site direct mutagenesis protocol was adapted from [http://www.stratagene.com/manuals/200518.pdf Stratgene]. Oligos were designed to have a high (>78°C) Tm by applying the formula
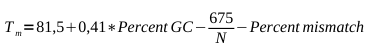 The following scheme was used for pipetting:
The following scheme was used for pipetting:
|
|
| 1 µl template (10 ng)
|
| 1 µl primer 1 (25µM)
|
| 1 µl primer 2 (25µM)
|
| 25 µl Mastermix 2x (Phusion)
|
| 22 µl dH2O
|
The PCR procedure was as follows:
- Initial denaturation, 1 cycle
- Amplification, 16 cycles
- denaturation 95 °C, 50 sec
- annealing, 60 °C, 50 sec
- extension, 72 °C, 40sec/kb + 30sec
- Final Extension,1 cycle
- 4 °C hold, forever
After completion, 1 µL of DpnI (NEB) was added and the mix was incubated for 1h at 37°C. Then, DH5alpha cells were transformed as described above.
DNA synthesis
Oligos were designed using [http://baderlab.bme.jhu.edu/gd/ GeneDesign]. 15Bp were chosen as an overhang and 56 °C as an annealing temperature. Oligos were ordered at a concentration of 100µM. 1:10 dilutions of the first and last oligo were prepared. Then, all oligos (including first and last) were pooled at 1µL each. Water was added to 10x the original volume. (So if a gene is synthesized from x = 14 oligos, water was added to 10*x = 140µL). This pool was then diluted 1:10. x µL of the dilution were put into a PCR reaction with 25µL Phusion master mix and 25-x µL water. PCR was conducted as following:
- Initial denaturation, 1 cycle
- Amplification, 7 cycles
- denaturation 95 °C, 30 sec
- annealing, 58 °C, 30 sec
- extension, 72 °C, 1 minute
- 4 °C hold, forever
Afterwards, 1µL of the 1:10 dilutions of the innermost and outermost primer were added. The same PCR protocol was then repeated, but with 25 instead of 7 cycles. PCR products were run on a 2% agarose gel and gel purified.
Purification of DNA from PCR reactions
PCR products were purified by the QIAquick PCR Purification Kit from Qiagen following the instructions of the Qiagen Handbook. To check the purity and amount of extracted DNA an aliquot was analysed using a NanoDrop.
Enzymatic hydrolysis of DNA by restriction enzymes
Restriction digest
Restriction digest of DNA was used for analysis of purified DNA form mini or maxiprep or for isolation of specific DNA fragments for further cloning. Analytical digestions were routinely conducted in 20 µl volume,preparative digestions were routinely conducted in 50 µl volume. In all digestions a minimum of 2 Units restriction enzyme(s) was used per microgram DNA. Optimised buffer conditions were secured by using NEB buffer system. The final reaction volume was achieved by adding H2Odest. The sample was incubated at optimal temperature for the restriction enzyme(s) (usually 37 °C). Analysis and preparation of DNA were either done by gelelectrophoreses (loading dye was added to the samples and they were loaded on a agarose gel) or alternatively PCR Purification (PCR Purification QIAquick Kit from Qiagen)
Phosphatasing with Shrimp Alkaline Phosphatase
SAP is used for Dephosphorylation of 5'-ends of DNA in Restriction Enzyme Reaction. After restriction digest 1 unit of SAP is added for every 1 pmol of DNA ends (about 1 μg of a 3 kb plasmid) and incubated at 37°C for 30-60 min. The reaction is stopped by heating at 65°C for 15 min (this completely inactivates SAP)
The minimum effective amount of SAP for dephosphorylation of 1 pmol of DNA termini in 1 hr at 37°C is:
- 0.05 units for 5'-protruding termini
- 0.05 units for blunt termini
- 0.1 units for 5'-recessed termini
Agarose gel electrophoresis for separating DNA
In the agarose gel electrophoresis a mixture of DNA fragments with different sizes are separated in an electrical field by their size. This is achieved by moving the negatively charged DNA through an agarose matrix while shorter fragments will run faster. The size of the pores can be controlled by agarose concentration. The higher the agarose concentration the smaller the pores are and the smaller fragments can be separated. Agarose concentrations between 0.7 and 1.5 % agarose in 0.5x TE buffer were used.
The agarose was dissolved completely by heating up and 0.1 µg/ml ethidium bromide was added.
The DNA fragments were separated using a constant voltage between 80 and 130 V. Under UV light (λ = 254 nm) DNA is visible through the unspecific intercalated ethidium bromide and can be documented or cut out and extracted from the gel.
Isolation of DNA fragments from an agarose gel
Plasmid DNA and DNA fragments were extracted using the Gel Extraction Kit from Qiagen following the manufacture instructions. To check the purity and amount of extracted DNA an aliquot was analysed using a NanoDrop.
Cell Culture
Splitting cells
Cultures of HeLa, MCF7 and U2OS cells are splitted 1:10 every 3-4 days or 1:3 every day: DMEM+++ (see Material) covering the cells is removed and the cells are washed with 3 ml PBS. The PBS is removed after the washing step and 3 ml of trypsin are added to the flask; almost all of the Trypsin is removed again until the cells are only slightly covered with Trypsin. The trypsinized cells are then incubated for 5 min at 37°C and 5% CO2. After this incubation time, the cells are detached from the bottom and the flask is filled with 5 ml of DMEM+++. Cells are resuspended in medium by pipetting up and down. To achieve a diluton of 1:10, 0,5 ml of the suspension is transferred to another flask and 5 ml of DMEM+++ are added. This dilution is then incubated for 3-4 days at 37°C and 5% CO2. The rest of the cell suspension (about 4 ml) is transferred to a 50 ml falcon tube and the same volume of DMEM+++ is added to the falcon. 9,6 µl of this dilution are transferred to a counting chamber and the number of cells is determined (x*10^4).
- Note 1: HeLa and U2-OS seem to grow faster than MCF-7; check notebook for information on splitting ratios
Transfection of Mammalian cells
1.Fugene (Roche) 10^5 cells were moved to 6-well plates. 2,5 mL of DMEM medium plus addiditives were added.The following day, 97 µL pure DMEM was incubated for 5 minutes with 3µL Fugene6 reagent at roomt temperature. Afterwards, the mixture was added dropwise to 500ng of plasmid and allowed to incubate for 15 minutes at RT. The mixture was then added dropwise to the cells.Cells were incubated for 24 hours without selection pressure, afterwards, medium was changed and Zeomycine was added to a concentration of 350ng/mL
2. Effectene Transfection (Qiagen) Move x (for apropriate amounts see table below) cells to 6-well plates/ LabTek 8 chamber and add 2 ml/0,5 ml DMEM+++. Incubate over night at 37°C and 5% CO2.The following day, dilute x µg of DNA in Buffer EC to a total volume of x µl (note: Yara just adds 100 µl/50 µl). Add x µl of Enhancer and mix by tapping the tube (or vortex for 1 sec). Incubate for 2-5 min at RT. Add x µl of Effectene Transfection reagent to the DNA-Enhancer Mixture. Mix by tapping the tube (or vortexing for 10 s or pipetting up and down 5 times). Incubate samples for 5- 10 min at RT to allow for transfection-complex formation. While complex formation takes place, aspire medium and add x ml of fresh growth medium. After incubation time add the transfection
-complexes drop-wise to the cells on the well. Gently swirl the plate to ensure uniform distribution of the complexes.
- transient transfection: remove Effectene-DNA complexes after 6-18 hours, wash cells 1x with sterile PBS, add fresh medium. Incubate cells with the complexes under normal growth conditions for an appropriate time for expression.
- stable transfection: change medium after 24 hours (wash with PBS); check confluency after 48 hours - (if required) split to 25% confluency, add fresh medium and incubate 2-3 hours until cells have attached to culture dish. Then, remove medium and add fresh DMEM+++/Zeocin.
| Culture format
| Number of cells
| DNA [µg]
| Enhancer [µl]
| Final Volume of DNA in Buffer EC [µl]
| Effectene reagent [µl]
| Volume of Medium to add to cells [µl]
|
| 96-well
| 10^4
| 0,1
| 0,8
| 30
| 2,5
| 100
|
| Ibidi
| 1,5*10^4
| 0,15
| 1,2
| 50
| 4
| 200-250
|
| 24-well
| 4*10^4
| 0,2
| 1,6
| 60
| 5,0
| 700
|
| 6-well
| 10^5
| 0,4
| 3,2
| 100
| 10
| 2000
|
Freeze cells
Cells to be frozen should be confluent, but not too dense. Medium covering the cells is removed and cells are washed once with PBS. Afterwards cells are rinsed with 3 ml Trypsin; Trypsin is removed until the cells are only slightly covered (about 1 ml) and cells are incubated for about 5 min. After incubation, cells are resuspended in medium (at least about 4-5 times as much as Trypsin to inactivate that; used 5 ml). The suspension is transferred to 15 ml falcon and centrifuged at 2000 rpm for 3-5 minutes. The supernatant is discarded and the remaining pellet is resuspended in DMEM+++ (here: 7,5 ml for 5 aliquots, 1,5 ml each). 1,5 ml of the suspension is distributed into freezer vials. After adding of 150 µl DMSO to each vial, cells are put on ice and stored at -80°C.
Picking cells with cloning disks
Foci of cells, which were under selection pressure (zeocin) are marked on the bottom of the petri dish (use pen from underneath the microscope). As many cloning disks as foci available are prepared by incubating them 3-5 minutes in trypsin (in a petri dish). Medium covering the cells is removed and cells are washed with PBS. PBS is removed till the cells are only slightly covered with it. The cloning disks are placed on the previousely marked spots and incubated for 3 minutes at 37 °C. During incubation a 96-well plate is prepared with 0,7 ml DMEM+++/Zeocin per well. The clonig disks are picked up with pressure (or swipe a little) to remove the entire foci off the plate and the cloning disk is transferred to the 96-well plate (one disk per well). Medium is pipetted up and down to loosen the cells from the cloning disks.
Fix cells for microscopy
The medium is removed from the cells on cover slips. 500 µl of 4% Paraformaldehyde (in PBS) is added to the cells and incubated for 10 minutes. PFA is taken off and cells are washed with 500 µl of PBS. Slides are labeled and a small drop of fluoromount is added with a pipet tip. Cover slips are washed in dH20, by picking them up with tweezers and dipping them into the water. Afterwards cover slips are dried with filter paper without the paper touching the cover slip side containing the cells. Cover slips are layed down on the drop of fluoromount with the cells facing downwards. Fixed cells are dried overnight and can then be used for fluorescence microscopy.
Flow Cytometry (FC)
Before preparing samples for flow cytometry the flow cytometer has to be turned on at least 20 min before the run.
Samples are then prepared in the following way (this is for a 96 well plate): As much DMEM+++ as possible is removed from the cells. 60 µl Trypsin are added and the cells are incubated at 37 C for 10-15 min. Then, 160 µl of 1xPBS + 1% BSA are added. Samples are now ready for FC. At the FC the correct plate format is chosen. The plate is placed in the machine and the lid is taken off. Measurement parameters such as plateformat, wells, and the desired protocol are chosen from the list. Volume and Mix iterations are are also chosen and saved as... .The discriminator is set to increase the threshold(2-10). The measurement is started with forward vs. side scatter in order to find cells. When cells are found, a gate (line) is added around the cells or cell candidates. Selected cells are then measured.
Induction
To induce the respective pathways specifically, we searched literature and used knowledge of groups working with the drugs. The following table is an overview of the pathways, the used drugs and the conditions for activation.
| Pathway | mode | cell type | drug | stock concentration | dilution | final concentration | medium | time to leave on cells | remarks |
|
| HIF [1] | activator | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=Product%20No.%7CBRAND_KEY&N4=D9533%7CSIGMA&N25=0&QS=ON&F=SPEC DFO] | 50 mM | 1:500 | 100 μM | minimal medium | 24 h | together with HIF-bag |
|
| | inhibitor | MCF-7 | Everolimus | | 1:500 000 | | minimal medium | 24 h | without HIF-bag |
|
| SREBP [2], [8] | activator | HeLa | Lipid-depleted-Serum (LDS) | | 1:200 | | DMEM++ | 2-3 days | put on cells after transfection and put back on after 3 h with HPCD |
|
| | activator | HeLa | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=H107%7CSIGMA&N25=0&QS=ON&F=SPEC HPCD] | | 1:100 | | DMEM++ with LDS | 3 h | add after cells were 2-3 days in DMEM++ +LDS |
|
| | inhibitor | HeLa | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=C8667%7CSIGMA&N25=0&QS=ON&F=SPEC Cholesterol] | | | | DMEM+++ | 2-3 days | |
|
| | inhibitor | HeLa | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=L7914%7CSIGMA&N25=0&QS=ON&F=SPEC LDL] | | | | DMEM+++ | 2-3 days | |
|
| NFκB [1] | activator | U2-OS | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?N4=T0157%7CSIGMA&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&F=SPEC&lang=null%3E TNFα] | 2,5 mM | 1:1000 | 2,5 μM | DMEM++ | 10 h | |
|
| p53 [1],[5] | activator | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=C9911%7CSIGMA&N25=0&QS=ON&F=SPEC Camptothecin] | 2 mM | 1:1000 | 2 μM | minimal medium | 12 h | |
|
| | inhibitor | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=P4359%7CSIGMA&N25=0&QS=ON&F=SPEC Pifithrin-α] | 2 mM | 1:1000 | 2 μM | minimal medium | 12 h | |
|
| AHR [6] | activator | HeLa | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?N4=B1760%7CSIGMA&N5=Product%20No.%7CBRAND_KEY&F=SPEC Benzo-a-pyrene] | | 1:10 000 | | DMEM+++ | 18 h | |
|
| | activator | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?lang=en&N4=48599%7CSUPELCO&N5=SEARCH_CONCAT_PNO%6CBRAND_KEY&F=SPEC TCDD] | | 1:3000 | | DMEM++ | 48 h | |
|
| PPARγ [3],[4] | activator | U2-OS | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?N4=88405X%7CFLUKA&N5=Product%20No.%7CBRAND_KEY&F=SPEC 2,4-Thiazolidinedione] | 500 mM | 1:100 000 | 5 μM | DMEM+++ | 60 h | |
|
| Estrogen receptor | activator | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?N4=E8875%7CSIGMA&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&F=SPEC β-Estradiol] | 100 mM | 1:10 000 000 | 10 μM | DMEM++ | 24 h | keep cells with DMEM++ for 24 h after transfections, then add drug |
|
| VDR [7] | activator | MCF-7 | [http://www.sigmaaldrich.com/catalog/ProductDetail.do?D7=0&N5=SEARCH_CONCAT_PNO%7CBRAND_KEY&N4=D1530%7CSIGMA&N25=0&QS=ON&F=SPEC Vitamine D3] | 100 μM | 1:1000/ 1:10000 | 100 nM/ 1 nM | DMEM+++ | 24 h, 48 h, 72 h, 96 h | TECAN read every 24 hours -> see induction: FACS |
|
| c-Jun [1] | activator | HeLa | EGF | 100μg/mL | 1:1000 | 100ng/mL | | | |
|
RA-PCR
For a RA-PCR protocol, please refer to Project page
We used the following Oligo concentrations:
| p53
| NFκB II
| HIF
| Activator Mix
|
6µL p53 (O.91)
5µL p53 reverse (O.188)
1µL random (O.56)
1µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 3µL NFkB-1 (O.93)
3µL NFκB-2 (O.94)
4µL NFκB-rev (O.194)
3µL Random (O.56)
2µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 2,5µL HIF-1 (O.53)
2,5µL HIF-2 (O.54)
1µL CREB(O.89)
3µL HIF-rev (O.189)
3µL Random (O.56)
1µL Stop 5 new (O.187)
1µL Stop 3 (O.58)
.2µL each Ap1, Sp1 (O.55, O.57)
| 2µL each Ap1, Sp1, CREB (O.55, O.57, O.89)
1µL each NFY, Empty (O.90, O.95)
Water to 30µL
|
| SREBP
| AHR
| pPARγ
| Estrogen receptor
|
5µL SREBP (O.208)
4µL SREBP reverse (O.209)
1µL Sp1 (O.57)
2µL random (O.56)
1µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 5µL AHR (O.212)
4µL AHR reverse (O.213)
2µL random (O.56)
1,5µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 5µL pPARγ (O.210)
4µL pPARγ reverse (O.211)
2µL random (O.56)
1,5µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
| 5µL Estrogen receptor (O.210)
4µL Estrogen receptor reverse (O.211)
2µL random (O.56)
1,5µL Activator Mix
0.8µL Stop 5 new (O.187)
0.8µL Stop 3 (O.58)
|
Measurement and Screening
Screening by TECAN
Cells were grown in black 96 well plates (PerkinElmer) and induced as described for the various pathways. Outer wells were left free. After induction, media was removed and replaced by PBS. Fluorescence was then measured using a TECAN infiniteM200 automated plate reader (excitation = 488nm, emission=518nm). In order to account for variations in cell number, cells were stained by [http://www.sigmaaldrich.com/etc/medialib/docs/Sigma/Product_Information_Sheet/b2261pis.Par.0001.File.tmp/b2261pis.pdf Hoechst 33342 dye] . PBS was removed and replaced by PBS containing Hoechst 33342 dye at a concentration of 1µg/mL. Cells were allowed to incubate at room temperature for 30 minutes. Afterwards, medium was replaced by PBS again and fluorescence (excitation = 355nm, emission=455nm) was measured again by TECAN. Fluoresences values from the first reading were divided by fluorescence values of the second reading and multiplied by the lowest value of the second reading. Clones showing induction were considered further.
Flow Cytometry
Cells were prepared in 96-well format with 10^4 cells/well and transfected with the promoter of interest and if necessary induction drug was added for inducible promoters. Before measurement, the medium was removed, the cells were washed with 1xPBS and trysinized with 60 μl of trypsin per well. After 10 minutes incubation at 37°C 1xPBS + 1% BSA were added up to a volume of 200 μl per well. When starting the flow cytometry measurement we first adjusted the gates and the background signal to the negative control (Fig. ?). Figure blablablu shows an exapmle of a gated positive control (JeT), where we can see a second normal distribution peak indicating the gated GFP positive cells.
Microscopy/ Image Analysis
Microscopy images of our samples fixed with 4% formaldehyde were taken using the Nikon Eclipse 90i upright automated widefield microscope in the Nikon Imaging Center at the University of Heidelberg. Each image was taken in the GFP and mCherry channel and the exposure time had to be adjusted for over- or underexposed images. The conversion factor was experimentally determined by imaging a fluorescent plate with exposure time varying from 10 to 70 ms (the estimated range of our promoters) and plotting the mean grey values of these images versus time. A linear relationship between the exposure times and grey values was observed for this time frame and the conversion factor was determined to be 0.998. For image analysis we used the ImageJ (Image Processing and Analysis in Java) software measuring the mean grey values of each cell containing the promoter of interest. ImageJ is an open source software developed at the National Institutes of Health (http://rsbweb.nih.gov/ij/). The measurement is performed by setting the boundaries around the cells using the threshold in the mCherry image and generating a mask that is then applied to the image of the GFP channel in the following steps:
- Open image and select split channels
- Select mCherry image (all images should be 8-bit)
- Go to Image> Adjust> Threshold
- Set lower bar to maximum and adjust upper bar so that the cells are still visible
- Remove unwanted spots such as dead cell using the drawing tools
- Apply the threshold (image should appear in all white now with black spots indicating the cells)
- Go to Process> Math> Divide and divide the image by 255 (image should be all white now)
- Next go to Process> Image Calculator and multiply the GFP channel with the mCherry channel pictures.
- Now select the result image and adjust the threshold so that the lower bar is set to the maximum (255) and the upper bar is set to 1 (only GFP containing cells should be visible through the mask)
- Go to Analyze> Set Measurement and make sure the measurement is limited to the threshold and the result image of the multiplication is selected.
- Go to Analyze> Measure.
- Mean grey values should show up in the 'Results' window
Alternatively the images can be analyzed using the ROI manager where the cell have to be selected by hand using the drawing tools and added to the ROI list. In comparison the threshold method is much quicker and the results are comparable. We preselect the mCherry positive cells which are very likely to contain GFP. The mean grey values of 5-10 images per sample were averaged and adjusted to the same exposure time. The results were given in REU relative to JeT, which was always prepared as a reference sample for each measurement.
References
[1] Brady N. Bioquant Heidelberg, Eils group; Personal communication.
[2] Schilde J. Institute for human genetics at the University of Heidelberg; Personal communication.
[3] Panigrahy D., Singer, S., Shen L. Q., Butterfield C. E., Freedman D. A., Chen E. J., Moses M. A., Kilroy S., Duensing S., Fletcher C., Fletcher J. A., Hlatky L., Hahnfeldt P., Folkman J., Kaipainen A. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. Journal of clinical cancer 110: 923-932 (2002)
[4] Lucarelli E., Sangiorgi L., Maini V., Lattanzi G., Marmiroli S., Reggian, M., Mordenti M., Gobbi G. A., Scrimieri F., Bertoja A. Z. & Picci P. Troglitazione affects survival of human osteosarcoma cells. International Journal of Cancer 98 (3): 344-351 (2002)
[5] Komarov,P. G., Komarova E. A., Kondratov R. V., Christov-Tselkov K., Coon J. S., Chernov M. V., Gudkov A. V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285(5434):1733-7 (1999)
[6] Wang S. H., Liang C. T., Liu Y. W., Huang M. C., Huang S. C., Hong W. F., Su J. G. Crosstalk between activated forms of the aryl hydrocarbon receptor and glucocorticoid receptor. Toxicology. 262(2):87-97 (2009)
[7] Diesing D., Cordes T., Fischer D., Diedrich K., Friedrich M. Vitamin D--metabolism in the human breast cancer cell line MCF-7. Anticancer Res. 26(4A): 2755-9 (2006)
[8] Bartz F., Kern L., Erz D., Zhu M., Gilbert D., Meinhof T., Wirkner U., Erfle H., Muckenthaler M., Pepperkok R., Runz H. Identification of Cholesterol-Regulating Genes by Targeted RNAi Screening. Cell Metabolism 10: 63-75 (2009)
[9] Runz H., Miura K., Weiss M. & Pepperkok R. Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. The EMBO Journal 25: 2953 - 2965 (2006)
|
|
 "
"
