|
|
CAROL
Second Attempt to Plasmid Isolation
- Spun the cells down from overnight cultures and froze cells overnight for the weekend.
|
|
|
CHINMOYEE
Three Gene Repressilator Presentation
Presented Ander's Modeling examples of the Three Gene Repressilator .
Looked at the Optimization Toolbox.
|
|
|
EMILY
Friday Team Meeting
- Had a team meeting where each subproject presented their recent work. Lab group talked about verification of genes of interest in TOPO vectors, BBK amplification PCR and cloning into Biobrick vectors (psB1AC3 and psB1AK3).
|
|
|
FAHD
Team Meeting for July 12th 2009
Today we had our team meeting and I presented on behalf of the Human Practices aspect of our project
|
|
|
IMAN
Writing and Learning Functions
As Afshin was away for this week, I worked on various functions involved in the model namely “trippleNumbers”, “calProbibility”, “applyRule”, “updateEnv”, and “simulate”. The first two functions get the information from each membrane and send the information to Gillespie’s algorithm. Then this algorithm chooses a rule which is most likely to occur. The chosen rule applies to the system using “applyRule” function. Then we need to update the system, and inform the other membranes about the rule applied to the system. It is the responsibility of “updateEnv”. I had two meetings this week as well, one with iGEM members and the other one with Lindsay members. I worked on the kidney model for Lindsay project, which I am not going to explain about it here.
|
|
|
JEREMY
Gel of BBK Amplification of LuxPQ
A 0.8% agarose gel was made and the gradient PCR products (see June 11) were ran at 90V. The gel below shows the results, with wells 1-12 having luxPQ template and well 13 being the negative control (water). We can see that all PCR products are of the correct size (~3.8kb = luxPQ).
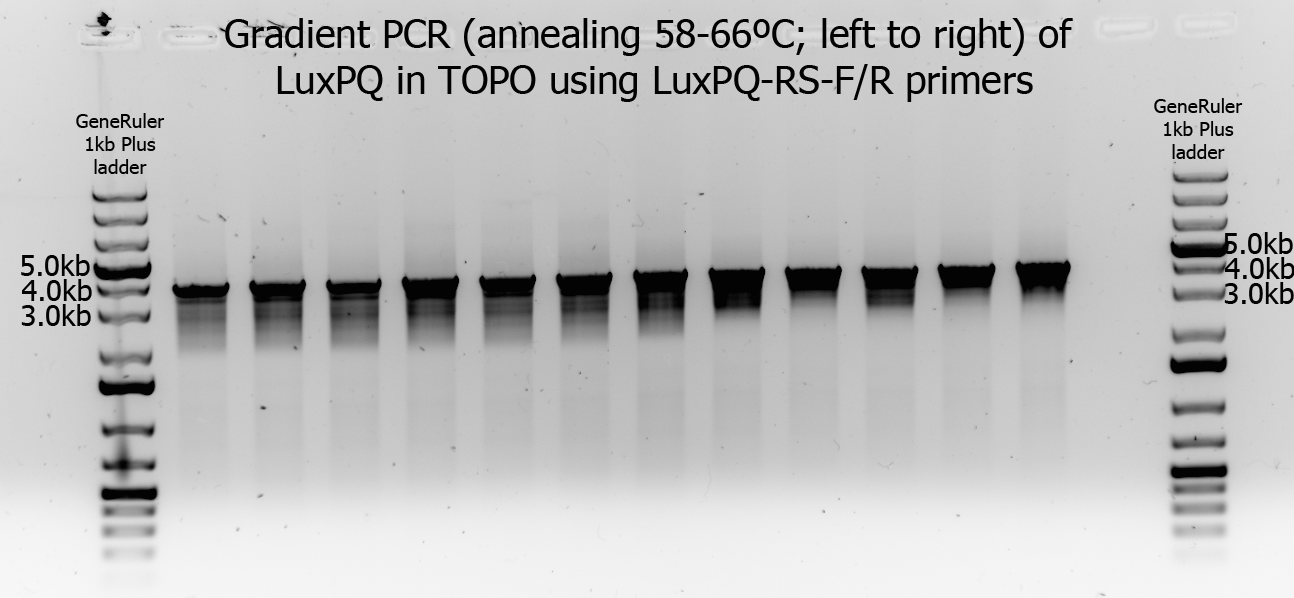
|
|
|
KEVIN
Team meeting
No lab experiments were performed.
|
|
|
MANDY
Second Life Brainstorming
While continuing my writing of notecards for gel prep, gel electrophoresis, and LB plate prep, we had a mini-SL team meeting in order to list the components we wanted to teach and thus what we wanted to build so we could check things off the list in order to ensure that we completed as much of it as possible by the end of the summer.
|
|
|
PATRICK
Stay Put
Retrospective Notebook: This entry was not written on this day, but derived later from working notes I made that day.
Began working on a newer 'stay put' script for the objects, this one changing their physics status when the object is clicked. With physics off, an object hangs frozen in space, and can intersect with other objects without difficulty. With physics on, an object can be dragged around with the mouse, and be made to collide with other objects (but cannot occupy the same space as another!) By default, objects would be nonphysical, and only set to physical when the user needs to move them around. The end result is that all of the objects are *much* less twitchy and glitchy, as the physics engine often makes physical objects pop around.
Began planning out just what material we would need to present within the sim, to get users up to speed for using the biobricks.
|
|
|
PRIMA
Shadowed Vicky with her Restriction digest
We had a team meeting where each iGEM subdivision presented their work for the week.
I shadoewd Vicky wither her Restriction Digest today. Although I did not quite understand her part completely, I tried my best to follow through the procedures and to understand why she was doing what she was doing.
Procedure for RD:
oth the vector and the insert need to have their own separate tube, at least in the beginning. This is important because it allows for clean addition new parts to a the circut
In the Insert Tube...
-600ng of DNA (To figure out the volume, the calculation is 600 / concentration of plasmid. This gives you volume in μL).
-Water, so that the volume of DNA and water in the tube is 35 μL
-4 μL of React 1 Buffer
-0.5 μL of EcoR1
-0.5 μL of Spe1
In the vector Tube...
-250ng of DNA (To figure out the volume, the calculation is 250 / concentration of plasmid. This gives you volume in μL).
-Water, so that the volume of DNA and water in the tube is 35 μL
-4 μL of React 2 Buffer
-0.5 μL of EcoR1
-0.5 μL of Xba1
Put both tubes into the 37°C water bath for one hour. After, place them into the 65°C heating block for 10 minutes. This destroys any enzymes in the tube (which is ok, because by now they’ve done all they need to). Take the insert out, and put it in a -20°C freezer.
|
|
|
STEFAN
More things added into the Synthetic Kingdom
Added a bunch of new bacteria and replicated them...the place is now teeming with life. Started brainstorming some interactive applications of synthetic biology I could showcase.
|
|
|
VICKI
Purification of gradient PCR product
Purpose: to purify the LuxOD47A BBk that was produced yesterday in the gradient PCR
Protocol: Please refer to the protocol page. Concentrations were measured after the product was purified.
Restriction digest of LuxOD47A BBk (insert) and psB1AC3 (vector)
Purpose: The LuxOD47A BBk DNA from yesterday’s gradient PCR is in linear form. This will prepare the ends so that it can be inserted into a BioBrick vector.
Protocol:
This is the restriction digest component of the construction protocol, which we describe on our protocol page. We attempted the construction with 2 different sets of enzymes: 6 tubes with cuts at XbaI and PstI , and 6 tubes with cuts at EcoRI and PstI. Both were prepared in REact 2 buffer. Once prepared, the tubes were left in the water bath at 37 degrees C for 6.5 hours; heatshocked at 65 degrees C on a heating block to deactivate the restriction enzymes in the tube; and placed in the -20 degrees C freezer.
|
 "
"











