 "
"
Orthogonal coiled-coils.html
From 2009.igem.org

|
|
De novo design of orthogonal coiled-coil forming segments
In this way selected system of coiled-coil forming segments is called orthogonal. At the N-terminal segment of each coiled-coil segment we introduced a Ser-Pro-Glu-Asp tetrapeptide, which is composed of contributions of the preferred residues at the N-termini of coiled-coil helices (Sun et al., 2000; Cochran et al., 2001; Penel et al.,l 1999). Side chain of those residues can form hydrogen bond with polypeptide backbone of the residues in the fist turn, interact with a dipole moment of helix and have conformational properties compatible with the beginning of a helix and at the same time serve to prevent formation of an extended helix, that might form between the consecutive coiled-coil forming segments.
To predict coiled-coil interactions we examined two algorithms (see Hagemann et al., 2009; Fong et al., 2006) and in the end chose to follow Hageman et al.:
There are many factors that affect stability: hydrophobic burial, propensity, solubility, electrostatic interaction of flanking residues and others. This algorithm considers three factors: core, electrostatic and propensity which seem to have the key role in CC formation.
The calculation is shown in following scheme (shown on a fictional protein pair)(Figure 1):
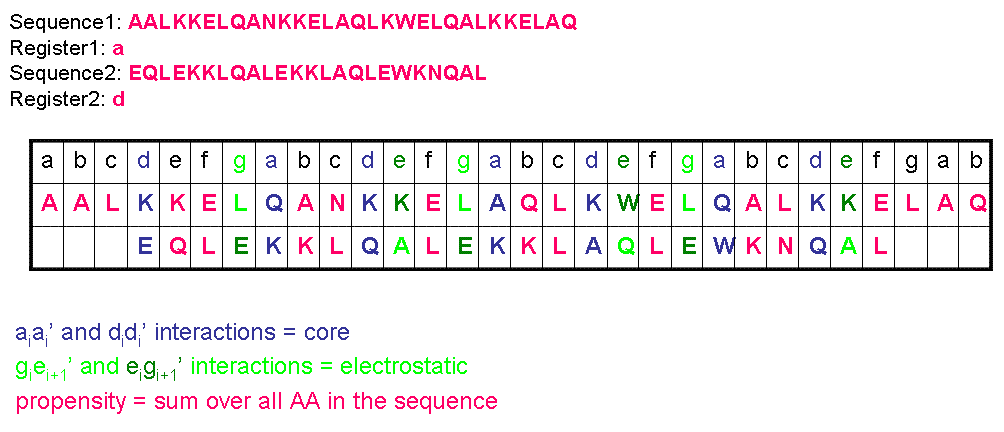
Figure 1 We extended this algorithm to pairs with different lenghts and register as well as antiparallel peptides - LINK Complete algorithm (in C, easily applicable to C++/Matlab/Mathematica): LINK and in Excel (just for pairs of with set register): LINK Identification of a set of orthogonal coiled-coil pairs
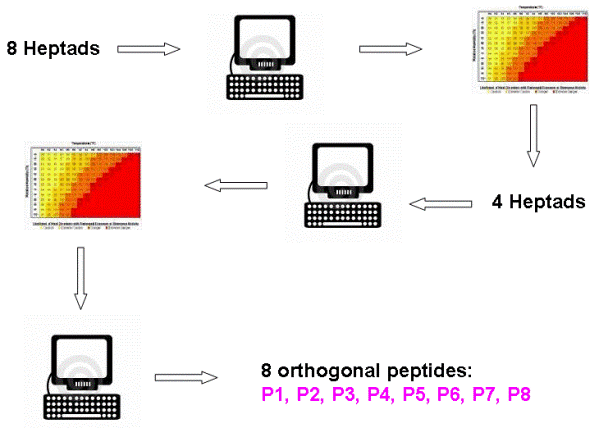
Figure 2 Figure 2 above shows our approach: First we calculated the interaction energy between all possible pairs of 8 different heptads. The interaction of coiled-coil segment is additive, therefore the interaction between larger coiled-coil segments, composed of several heptads could be deduced from the interacting contributions of all heptads. Next we generated a table where we calculated the interaction energy between all possible combinations of coiled-coil-forming segments composed of four heptads in both parallel and antiparallel orientation. Then we prepared another algorithm (download and abstract follow link HERE) to pick the best set of orthogonal pairs from the generated table which was selected to maximize the difference between the least stable desired pair and most stable undesired pair in the selected set. Predicted temperature difference between the least stable desired pair and most stable undesired pair was predicted to be more than 70 ºC. As a result of this approach we found a system of 8 orthogonal designed peptides: P1, P2, P3, P4, P5, P6, P7 and P8, which form four pairs: P1+P2, P3+P4, P5+P6 and P7+P8. In the following chart we can see theoretically predicted melting temperatures (higher the temperature, the more stable the coiled-coil) 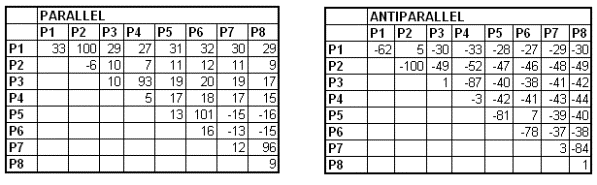
Figure 3 |