Notebook > AND Gate 1 > Input > Delete LVA tail from reverse tetR-tetP-GFP
Delete LVA tail from reverse tetR-tetP-GFP
Resource:
Reverse tetR-tetP-GFP: from Lin Min, plasmid. Renamed as TPG
Reverse tetR-tetP*2, (not very confirm): from Lin Min, plasmid, Renamed as TP1, TP2
Vector: a plasmid with Kan resistant. From Lin Min
Primer:
Delete LVA primer, with complement to 20 last bps of tetR coding sequence and a TAA+Xba1 tail. And it can be use with one of standard primers (Rev), to amplify any sequence between the end of tetR and standard suffix.
5’-GCTCTAGATTAGGACCCACTTTCACATTTAA-3’
Designed by myself.
2009.8.11
PCR: (helped by He Siheng)
| System | 20 uL |
|
| pfu enzyme | 1ul |
|
| primer | 1uL each | delete LVA primer and standard primer reverse one
|
| Buffer | 2 uL |
|
| water | 10uL |
|
| template TPG | 1uL |
|
| dNTP | 4uL |
|
Gel electrophoresis:
Products of PCR;
marker: 100bp 250bp 500bp 750bp 1kb 2kb 3kb 5kb
loading buffer and DNA dye: 6×
voltage and time: 60V 5min; 120V 15min
lane1: Marker;
lane2: product;
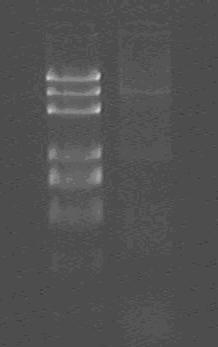
Obviously, PCR is failed!!!
Repeat!!
PCR: (repeat)
| System | 20 uL |
|
| pfu enzyme | 1ul |
|
| primer | 1uL each | delete LVA primer and standard primer reverse one
|
| Buffer | 2 uL |
|
| water | 10uL |
|
| template | 1uL | TP1, TP2, TPG
|
| dNTP | 4uL |
|
extending time 5 min;
2009.8.12
Gel electrophoresis:
Products of PCR;
marker: 100bp 250bp 500bp 750bp 1kb 2kb 3kb 5kb
loading buffer and DNA dye: 6×
voltage and time: 60V 5min; 120V 15min
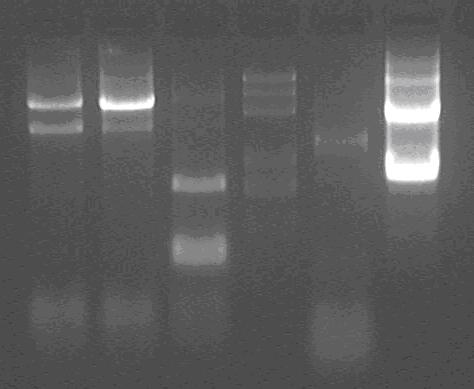
Lane1: TP1,
Lane2: TP2,
Lane3: TP1 (use another standard primer for one)
Lane4: Marker;
Lane5: negative control;
Lane6: TPG
The result is very strange:
TP1 and TP2 should be about 1kb, but there is not!
TPG should be about 2kB, but another 0.8k also very strong.
DNA Gel purification:
TPG (2kb) insert
Double digest:
TPG insert:
| Xba1 | 1uL
|
| Pst1 | 1uL
|
| DNA | 16uL
|
| Buffer | 2uL
|
Vector:
| Xba1 | 1uL
|
| Pst1 | 1uL
|
| plasmid | 4uL
|
| Buffer | 2uL
|
| water | 12 uL
|
37 ℃ 4 hour
Gel electrophoresis:
Products of digestion of vector
marker: 100bp 250bp 500bp 750bp 1kb 2kb 3kb 5kb
loading buffer and DNA dye: 6×
voltage and time: 60V 5min; 120V 15min
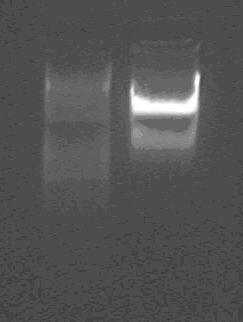
The vector is about 3kb.
DNA Gel purification:
Vector,
PCR product purification:
Products of digestion of insert (TPG)
DNA ligation:
| System | 10uL
|
| Insert | 6uL
|
| vector | 2uL
|
| buffer | 1uL
|
| T4 DNA ligase | 1uL
|
16℃ 4 hour
Insert: TPG;
Vector
Transformation:
Products of ligation, competent cells 50uL each,
Smear to LB plate with Kan.
2009.8.13
The plate (delete LVA TPG) is very well: more than 100 clones
And many colonies are become green under the blue light, which means that the expression of tetR can not fully repressed the promoter tetP.
The second picture is for comparison with no GFP colonies.

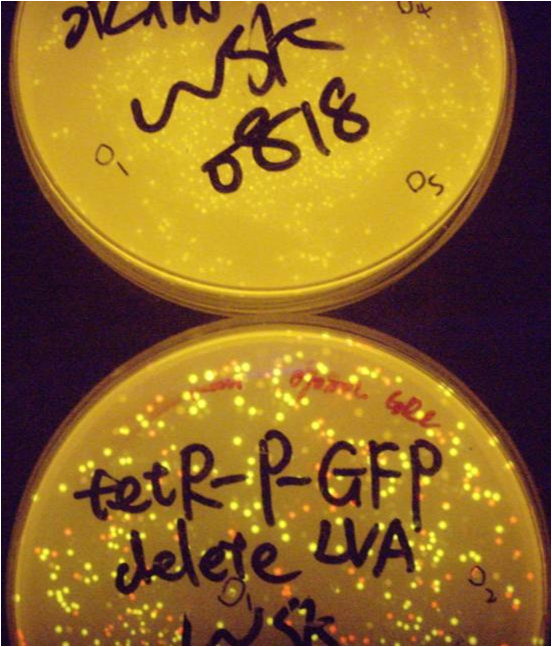
2009.8.14
Plasmid mini prep:
Del-LVA-TPG1, 2, 3.
Result
I successfully deleted the LVA from the Reverse tetR-tetP-GFP. Yet, the result is not very promising, because the colonies became green on the plate, without inducing.
It needs more quantitative data, but it is obviously that this cloning does not work very well.
^Top
|
 "
"





