"
"
Team:IBB Pune/Protocols
From 2009.igem.org
| (9 intermediate revisions not shown) | |||
| Line 2: | Line 2: | ||
{{Team:IBB_Pune/menu}} | {{Team:IBB_Pune/menu}} | ||
[[Image:PROTOCOLS.png|center|700px|center]] | [[Image:PROTOCOLS.png|center|700px|center]] | ||
| + | <br> | ||
| + | <br> | ||
| + | |||
<html> | <html> | ||
<span style="font-weight:bold; font-size:150%; color:#6600FF;">Maintenance of microbial cultures</span></html> | <span style="font-weight:bold; font-size:150%; color:#6600FF;">Maintenance of microbial cultures</span></html> | ||
| Line 25: | Line 28: | ||
Extraction of genomic DNA was done by the Chen and Kuo method with some modifications. | Extraction of genomic DNA was done by the Chen and Kuo method with some modifications. | ||
| - | + | [[Image:Klen.png|center|600px|center]] | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<html> | <html> | ||
<span style="font-weight:bold; font-size:150%; color:#6600FF;">Extraction of Plasmid DNA</span></html> | <span style="font-weight:bold; font-size:150%; color:#6600FF;">Extraction of Plasmid DNA</span></html> | ||
Extraction of plasmid DNA was done by the Birnboim and Doly (1979)method.The principle of the method is selective alkaline denaturation of high molecular weight chromosomal DNA while covalently closed circular DNA remains double-stranded. Adequate pH control is accomplished without using a pH meter. Upon neutralization, chromosomal DNA renatures to form an insoluble clot, leaving plasmid DNA in the supernatant. Large and small plasmid DNAs have been extracted by this method. | Extraction of plasmid DNA was done by the Birnboim and Doly (1979)method.The principle of the method is selective alkaline denaturation of high molecular weight chromosomal DNA while covalently closed circular DNA remains double-stranded. Adequate pH control is accomplished without using a pH meter. Upon neutralization, chromosomal DNA renatures to form an insoluble clot, leaving plasmid DNA in the supernatant. Large and small plasmid DNAs have been extracted by this method. | ||
| - | + | [[Image:Plasmidisolatn.png|center|700px|center]] | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<html> | <html> | ||
| Line 85: | Line 39: | ||
Agarose Gel Electrophoresis was performed on a horizontal gel apparatus for visualising DNA. Ethidium Bromide was used as the fluorescing dye. EtBr intercalates with the DNA strands and fluoresces under UV light thereby indicating the position of the band | Agarose Gel Electrophoresis was performed on a horizontal gel apparatus for visualising DNA. Ethidium Bromide was used as the fluorescing dye. EtBr intercalates with the DNA strands and fluoresces under UV light thereby indicating the position of the band | ||
| - | + | [[Image:Gelprot.png|center|700px|center]] | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<html> | <html> | ||
| Line 106: | Line 46: | ||
Genes to be amplified and identified by Polymerase Chain Reaction (PCR). | Genes to be amplified and identified by Polymerase Chain Reaction (PCR). | ||
| - | + | [[Image:Pcrfunnel.png|center|500px|center]] | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
[[Image:Protocoltable.png|center|400px|center]] | [[Image:Protocoltable.png|center|400px|center]] | ||
Latest revision as of 03:34, 21 October 2009

- Home
- Team
- Project
Summary Details Results Modeling
- Related
- Safety
- Parts
- Notebook
Maintenance of microbial cultures
Standard cultures used in our project are
1) E.coli DH5a
2) E.coli JM101
3) Acinetobacter baylyi BD413
4) Streptococcus pneumoniae R6
Glycerol stocks of all the strains are maintained at -80oC.
Strains are maintained on working plates for daily use, subcultured every week. Working plates used are Luria Bertani Agar for E.coli and Streptococcus strains and CLED (Cysteine Lactose Electrolyte Deficient) Agar for Acinetobactersp.
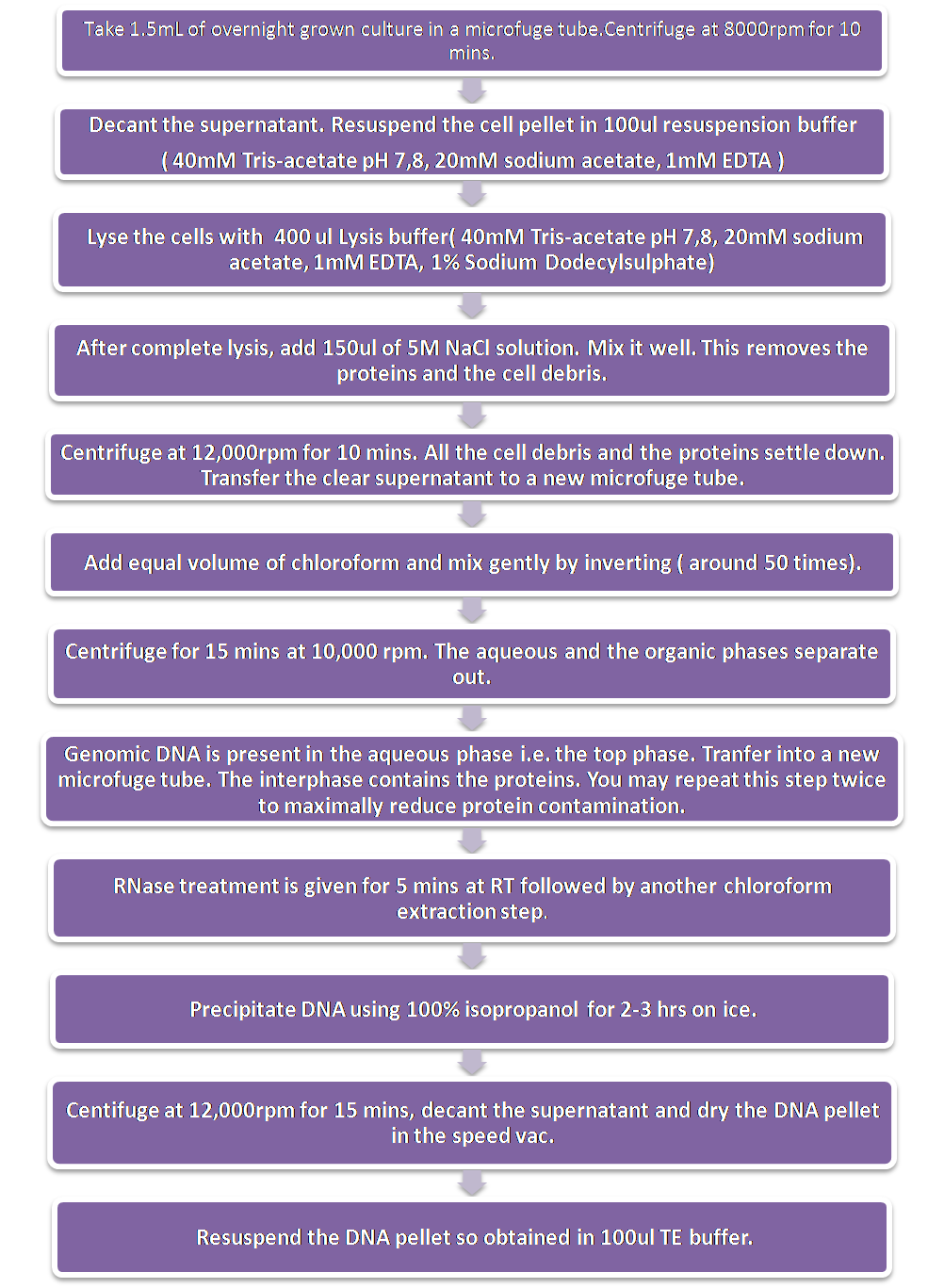
Extraction of Genomic DNAExtraction of genomic DNA was done by the Chen and Kuo method with some modifications.
Extraction of Plasmid DNA
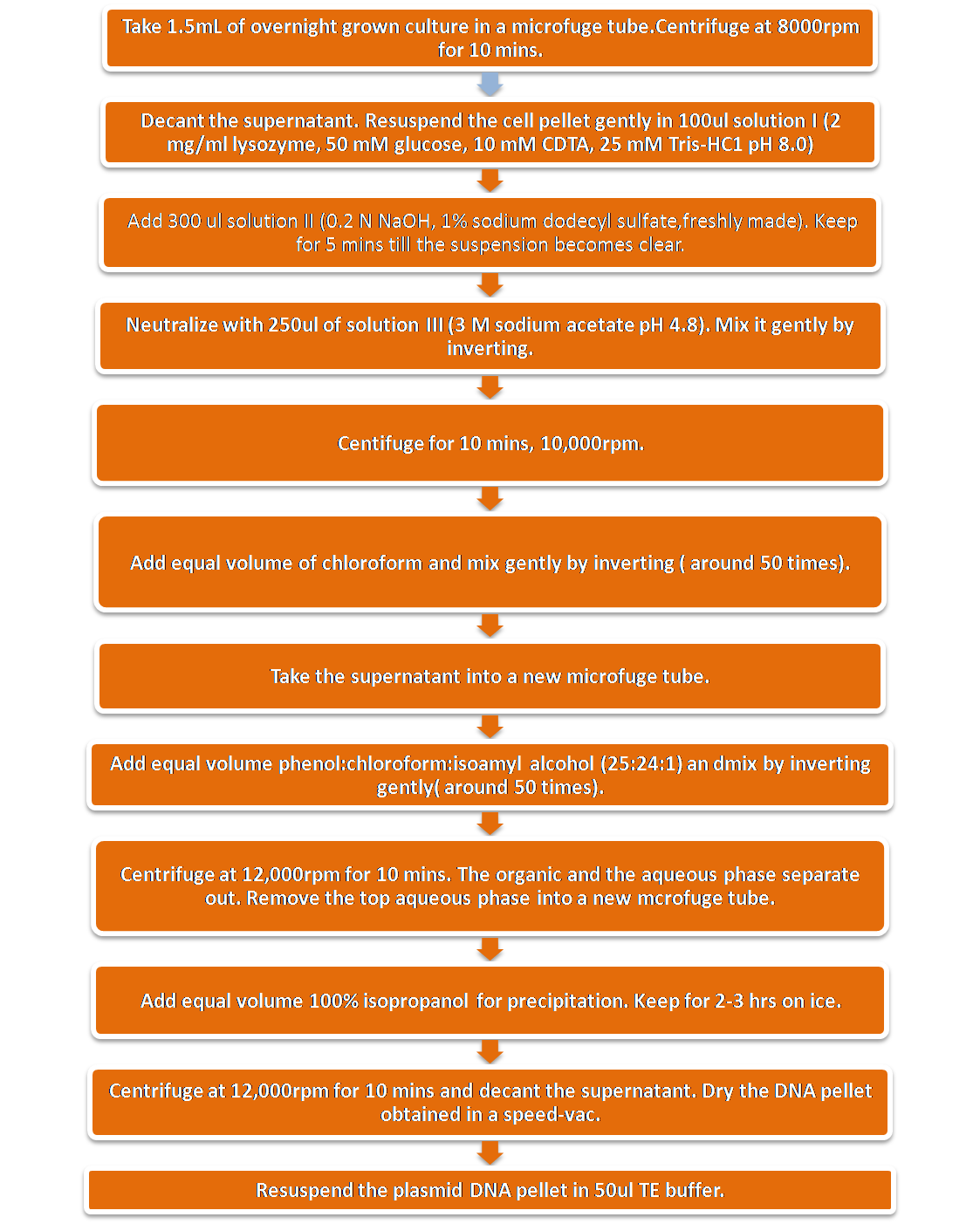
Extraction of plasmid DNA was done by the Birnboim and Doly (1979)method.The principle of the method is selective alkaline denaturation of high molecular weight chromosomal DNA while covalently closed circular DNA remains double-stranded. Adequate pH control is accomplished without using a pH meter. Upon neutralization, chromosomal DNA renatures to form an insoluble clot, leaving plasmid DNA in the supernatant. Large and small plasmid DNAs have been extracted by this method.
Agarose Gel Electrophoresis
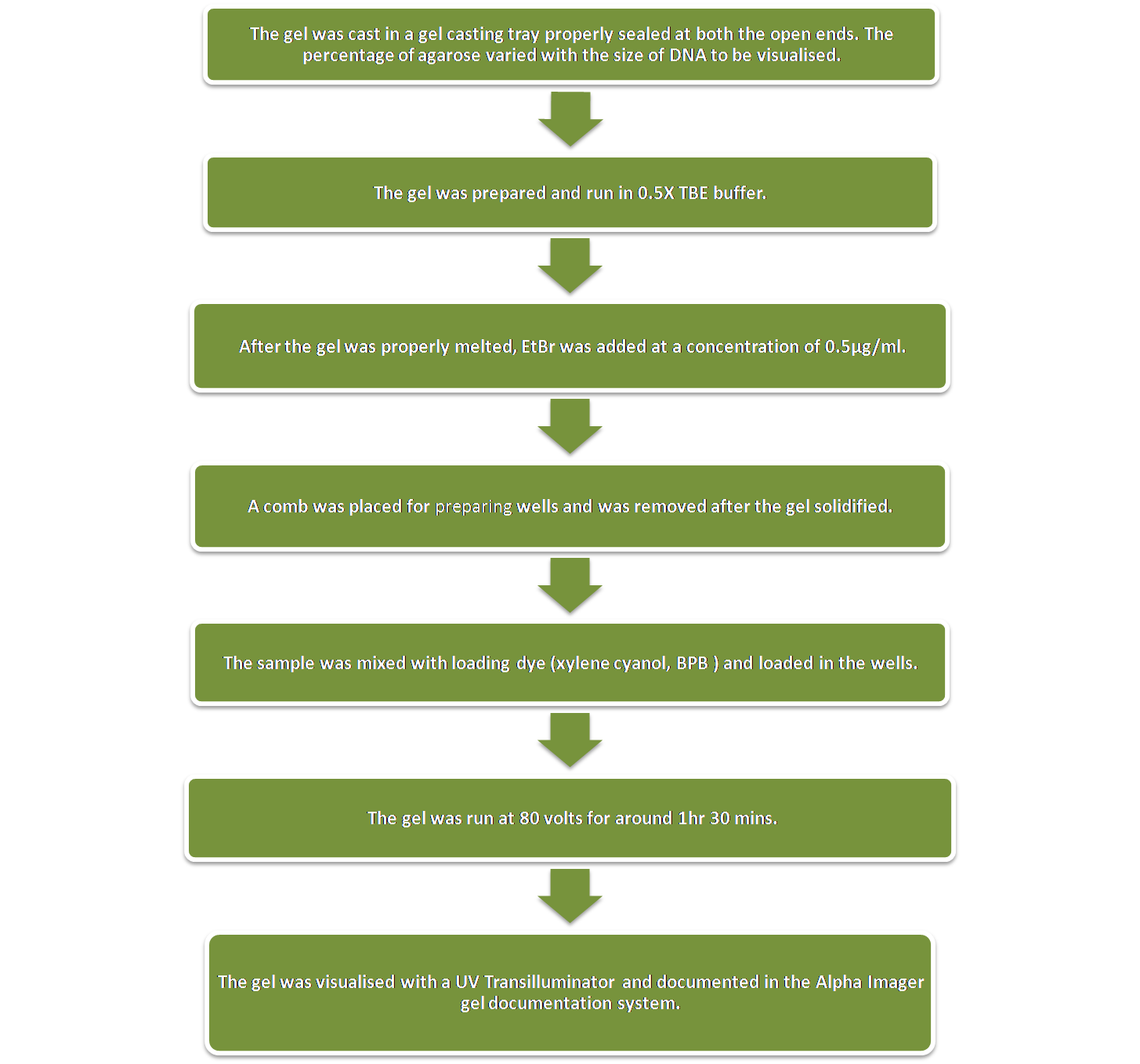
Agarose Gel Electrophoresis was performed on a horizontal gel apparatus for visualising DNA. Ethidium Bromide was used as the fluorescing dye. EtBr intercalates with the DNA strands and fluoresces under UV light thereby indicating the position of the band
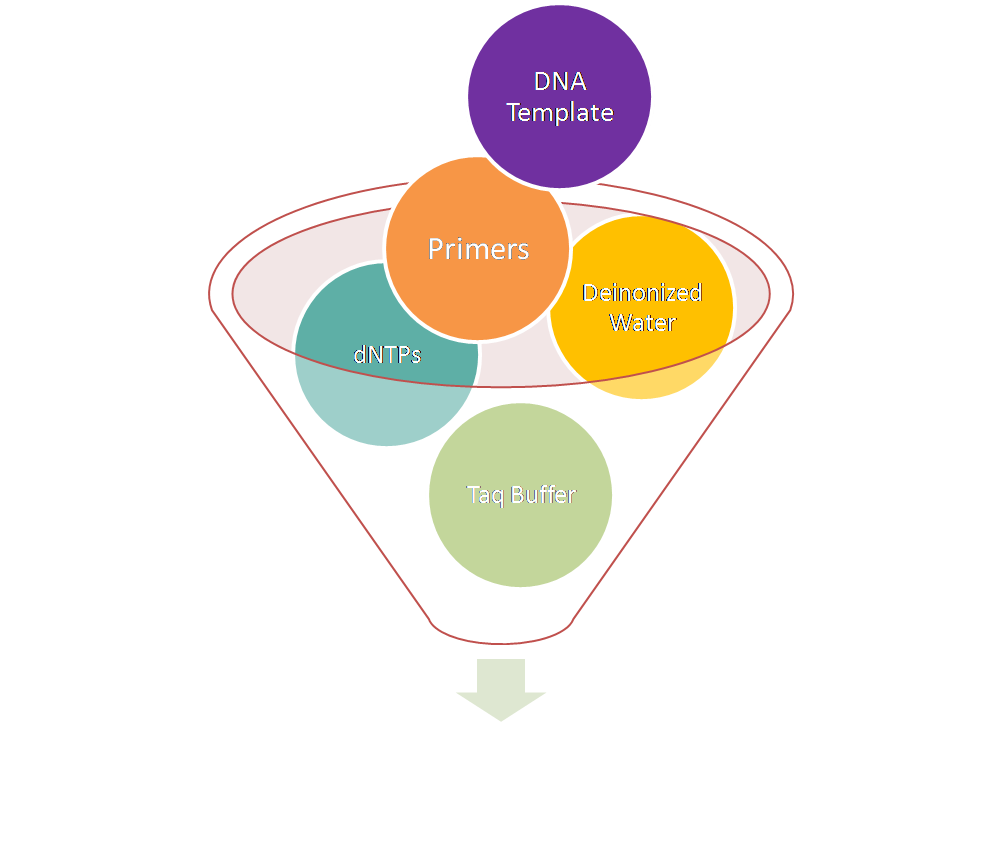
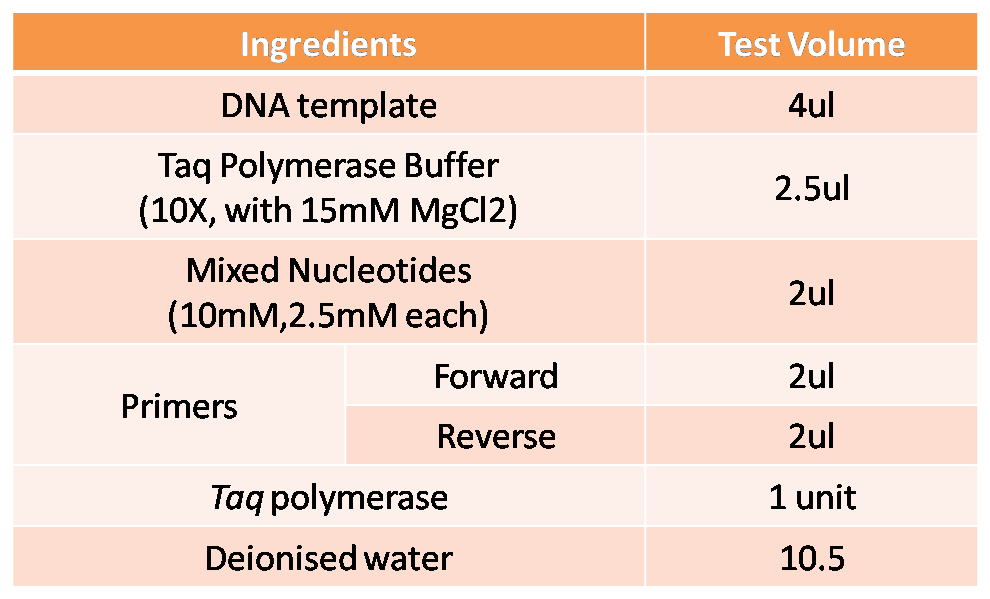
in vitro Gene Amplification
Genes to be amplified and identified by Polymerase Chain Reaction (PCR).