"
"
Team:IBB Pune/Protocols
From 2009.igem.org
| Line 2: | Line 2: | ||
{{Team:IBB_Pune/menu}} | {{Team:IBB_Pune/menu}} | ||
[[Image:PROTOCOLS.png|center|700px|center]] | [[Image:PROTOCOLS.png|center|700px|center]] | ||
| + | <br> | ||
| + | <br> | ||
| + | |||
<html> | <html> | ||
<span style="font-weight:bold; font-size:150%; color:#6600FF;">Maintenance of microbial cultures</span></html> | <span style="font-weight:bold; font-size:150%; color:#6600FF;">Maintenance of microbial cultures</span></html> | ||
Revision as of 15:12, 20 October 2009

- Home
- Team
- Project
Summary Details Results Modeling
- Related
- Safety
- Parts
- Notebook
Maintenance of microbial cultures
Standard cultures used in our project are
1) E.coli DH5a
2) E.coli JM101
3) Acinetobacter baylyi BD413
4) Streptococcus pneumoniae R6
Glycerol stocks of all the strains are maintained at -80oC.
Strains are maintained on working plates for daily use, subcultured every week. Working plates used are Luria Bertani Agar for E.coli and Streptococcus strains and CLED (Cysteine Lactose Electrolyte Deficient) Agar for Acinetobactersp.
Extraction of Genomic DNAExtraction of genomic DNA was done by the Chen and Kuo method with some modifications.
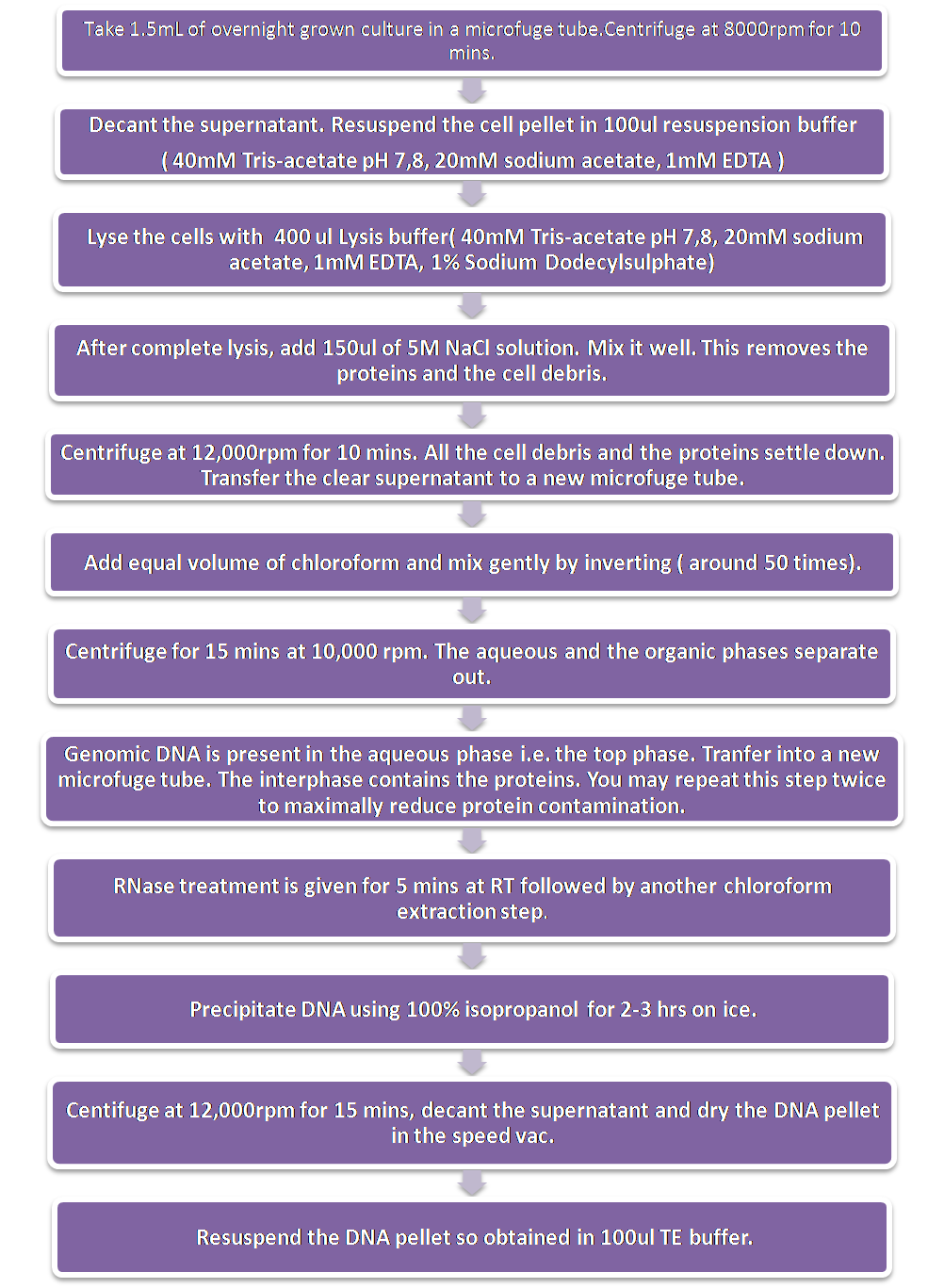
1) Take 1.5mL of overnight grown culture in a microfuge tube.Centrifuge at 8000rpm for 10 mins.
2) Decant the supernatant. Resuspend the cell pellet in 100ul resuspension buffer ( 40mM Tris-acetate pH 7,8, 20mM sodium acetate, 1mM EDTA )
3) Lyse the cells with 400 ul Lysis buffer( 40mM Tris-acetate pH 7,8, 20mM sodium acetate, 1mM EDTA, 1% Sodium Dodecylsulphate)
4) After complete lysis, add 150ul of 5M NaCl solution. Mix it well. This removes the proteins and the cell debris.
5) Centrifuge at 12,000rpm for 10 mins. All the cell debris and the proteins settle down. Transfer the clear supernatant to a new microfuge tube.
6) Add equal volume of chloroform and mix gently by inverting ( around 50 times).
7) Centrifuge for 15 mins at 10,000 rpm. The aqueous and the organic phases separate out.
8) Genomic DNA is present in the aqueous phase i.e. the top phase. Tranfer into a new microfuge tube. The interphase contains the proteins. You may repeat this step twice to maximally reduce protein contamination.
9) RNase treatment is given for 5 mins at RT followed by another chloroform extraction step.
10)Precipitate DNA using 100% isopropanol for 2-3 hrs on ice.
11) Centifuge at 12,000rpm for 15 mins, decant the supernatant and dry the DNA pellet in the speed vac.
12) Resuspend the DNA pellet so obtained in 100ul TE buffer.
Extraction of Plasmid DNAExtraction of plasmid DNA was done by the Birnboim and Doly (1979)method.The principle of the method is selective alkaline denaturation of high molecular weight chromosomal DNA while covalently closed circular DNA remains double-stranded. Adequate pH control is accomplished without using a pH meter. Upon neutralization, chromosomal DNA renatures to form an insoluble clot, leaving plasmid DNA in the supernatant. Large and small plasmid DNAs have been extracted by this method.
1) Take 1.5mL of overnight grown culture in a microfuge tube.Centrifuge at 8000rpm for 10 mins.2) Decant the supernatant. Resuspend the cell pellet gently in 100ul solution I (2 mg/ml lysozyme, 50 mM glucose, 10 mM CDTA, 25 mM Tris-HC1 pH 8.0)
3) Add 300 ul solution II (0.2 N NaOH, 1% sodium dodecyl sulfate,freshly made). Keep for 5 mins till the suspension becomes clear.
4) Neutralize with 250ul of solution III (3 M sodium acetate pH 4.8). Mix it gently by inverting.
5) Centifuge for 10 mins, 10,000rpm.
6) Take the supernatant into a new microfuge tube.
7) Add equal volume phenol:chloroform:isoamyl alcohol (25:24:1) an dmix by inverting gently( around 50 times).
8) Centrifuge at 12,000rpm for 10 mins. The organic and the aqueous phase separate out. Remove the top aqueous phase into a new mcrofuge tube.
9) Add equal volume 100% isopropanol for precipitation. Keep for 2-3 hrs on ice.
10) Centrifuge at 12,000rpm for 10 mins and decant the supernatant. Dry the DNA pellet obtained in a speed-vac.
11) Resuspend the plasmid DNA pellet in 50ul TE buffer.
Agarose Gel Electrophoresis
Agarose Gel Electrophoresis was performed on a horizontal gel apparatus for visualising DNA. Ethidium Bromide was used as the fluorescing dye. EtBr intercalates with the DNA strands and fluoresces under UV light thereby indicating the position of the band
1) The gel was cast in a gel casting tray properly sealed at both the open ends. The percentage of agarose varied with the size of DNA to be visualised.2) The gel was prepared and run in 0.5X TBE buffer.
3) After the gel was properly melted, EtBr was added at a concentration of 0.5μg/ml.
4) A comb was placed for preparing wells and was removed after the gel solidified.
5) The sample was mixed with loading dye (xylene cyanol, BPB ) and loaded in the wells.
6) The gel was run at 80 volts for around 1hr 30 mins.
7) The gel was visualised with a UV Transilluminator and documented in the Alpha Imager gel documentation system.
in vitro Gene Amplification
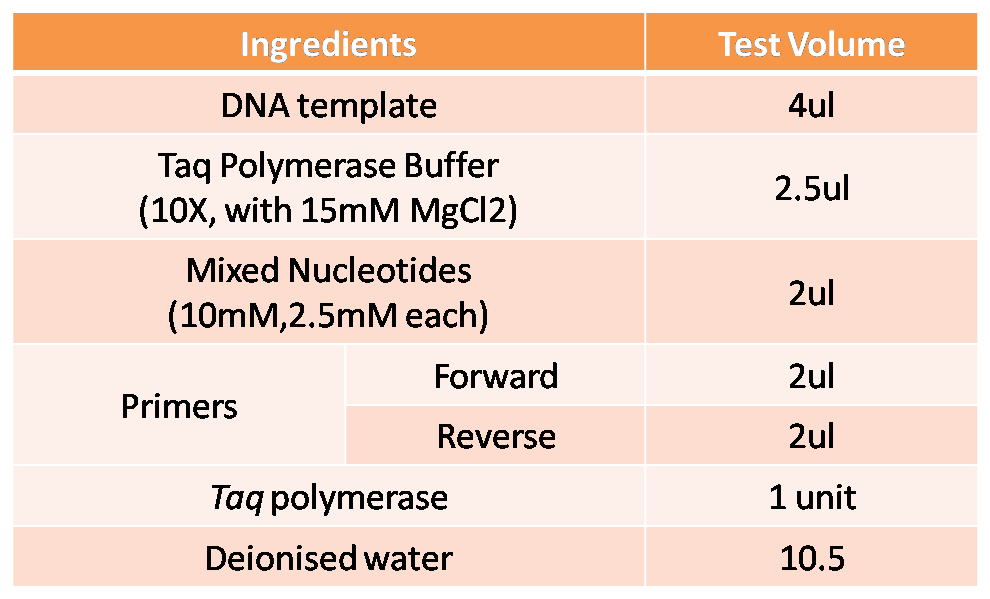
Genes to be amplified and identified by Polymerase Chain Reaction (PCR).
Chemicals and reagents:
- DNA template
- dNTPs(10mM, 2.5mM each)
- Taq Buffer (10X, with 15mM MgCl2)
- Primers
- Deionised water